Ruthenium-catalyzed intramolecular carbocyclization of alkynes with an sp3carbon involving an oxidative deprotonation process†
Bo-Xiao
Tang‡
b,
Ren-Jie
Song‡
a,
Cui-Yan
Wu
ab,
Zhi-Qiang
Wang
ab,
Yu
Liu
a,
Xiao-Cheng
Huang
ab,
Ye-Xiang
Xie
a and
Jin-Heng
Li
*a
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, China. E-mail: jhli@hnu.edu.cn; Fax: +0086731 8887 2531; Tel: +0086731 8887 2576
bKey Laboratory of Chemical Biology & Traditional Chinese Medicine Research (Ministry of Education), Hunan Normal University, Changsha, 410081, China
First published on 5th August 2011
Abstract
A novel RuCl3-catalyzed intramolecular oxidative hydration–deprotonation–cyclization method was developed for the synthesis of substituted quinolinones. The method serves as the first example of direct carbocyclization of an alkyne with an sp3carbon at the α-position of an amide through an oxidative deprotonation process.
Quinolinone and its derivatives are widely presented in numerous bioactive natural products, potent pharmaceutical drugs and various functional materials, making their efficient synthesis of great interest.1–6 These efficient strategies include the effective C–H bond functionalization method.4–6 However, a significantly more challenging task is to functionalize an sp3 C–H bond version of the reaction, since the reported examples involve only sp2 C–H bond functionalization methods, such as arylation of N-arylpropiolamides4 or N-(2-haloaryl)-benzamides5 (path a in Scheme 1) and amidation of 3-arylacrylamides (path b).6 We envisioned that quinolinones could be prepared from the carbocyclization of alkynes with an active sp3carbon at the α-position of a monofunctional carbonyl compound (path c).
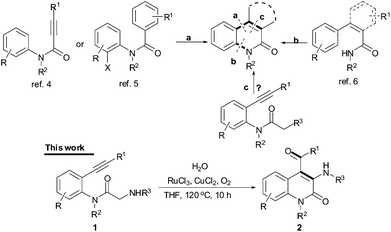 | ||
| Scheme 1 The C–H olefination and cyclization routes. | ||
Generally, activation of this α-position carbon in a monofunctional carbonyl system requires a strong base for deprotonation, leading to a carbon nucleophile or an enol intermediate for valuable organic targets.7 Here, we describe a base-free method for the synthesis of 3,4-disubstituted quinolin-2(1H)-one derivatives from ruthenium-catalyzed oxidative deprotonation and carbocyclization reactions of 2-acetamido-N-(2-ethynyl)aryl)acetamides (Scheme 1). To the best of our knowledge, it is the first example of direct carbocyclization of an alkyne with an sp3carbon at the α-position of an amide through an oxidative deprotonation process under neutral conditions.8
Ruthenium has proven to be highly efficient for the oxidative functionalization of sp3 C–H bonds in the presence of oxidants (often H2O2, t-BuOOH (TBHP) or O2).9,10 Thus, our investigation began with the reaction of 2-acetamido-N-methyl-N-(2-(phenylethynyl)phenyl)-acetamide (1a) under previously reported Ru/oxidant (TBHP or H2O2) conditions.9a–f These conditions were inert for the target reaction with a mixture of the C–N bonds decomposition products, which were determined by GC-MS analysis. Gratifyingly, the desired product, 2a, was observed in 9% yield under 10 mol % RuCl3 and 1 atm O2 conditions (entry 1, Table 1). We were pleased to find that CuCl2 could promote the reaction and the best results were observed at 20 mol % CuCl2 (entries 2–5). While 14% yield of 2a was isolated using 100 mol % CuCl2 oxidant alone (entry 2), 40 mol % CuCl2 at 1 atm of O2 enhanced the yield sharply to 55%, together with two other cyclization by-products 3a and 4a (entry 3). The yield increased slightly to 59% in the presence of 20 mol % CuCl2 and 1 atm O2 (entry 4). Notably, air was less effective (entry 6). Among the different reaction temperatures and amounts of RuCl3 examined, it turned out that 10 mol % RuCl3 at 120 °C afforded the best results (entries 7–11). It is noted that the reaction cannot take place without Ru catalysis (entry 9). Finally, another solvent, diglyme, was examined and the yield increased slightly (entry 12). The structure of 2a was unambiguously confirmed by the X-ray single-crystal diffraction analysis.†
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | RuCl3 (mol %) | [Cu] (mol %) | T/°C | Isolated yield (%) | ||
| 2a | 3a | 4a | ||||
| a Reaction conditions: 1a (0.3 mmol), H2O (6 equiv.), [Ru], additive, O2 (1 atm) and THF (3 mL) for 10 h. b In argon. c Air (1 atm) instead of O2. d Diglyme (3 mL) instead of THF. | ||||||
| 1 | RuCl3 (10) | — | 120 | 9 | trace | trace |
| 2b | RuCl3 (10) | CuCl2 (100) | 120 | 14 | trace | trace |
| 3 | RuCl3 (10) | CuCl2 (40) | 120 | 55 | 5 | 6 |
| 4 | RuCl3 (10) | CuCl2 (20) | 120 | 59 | 6 | 6 |
| 5 | RuCl3 (10) | CuCl2 (10) | 120 | 49 | trace | trace |
| 6c | RuCl3 (10) | CuCl2 (20) | 120 | 16 | trace | trace |
| 7 | RuCl3 (10) | CuCl2 (20) | 100 | 40 | 6 | 4 |
| 8 | RuCl3 (10) | CuCl2 (20) | 130 | 55 | 6 | 9 |
| 9 | — | CuCl2 (20) | 120 | 0 | 0 | 0 |
| 10 | RuCl3 (5) | CuCl2 (20) | 120 | 45 | trace | trace |
| 11 | RuCl3 (20) | CuCl2 (20) | 120 | 43 | trace | trace |
| 12d | RuCl3 (10) | CuCl2 (20) | 120 | 65 | 3 | 5 |
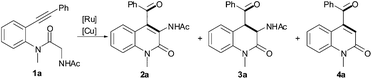
With the optimal reaction conditions in hand, a series of substrates with different functional substituents were examined (Table 2). We found that the analogous amides with the N-methyl group replaced by either a benzyl or an allyl group also successfully underwent the reaction with RuCl3, CuCl2 and O2 in moderate yields (2b–2c). However, substrates bearing an aliphatic n-propyl group in place of the acetyl group lost the activity dramatically (2d). Subsequently, several substituents with an R1group moiety at the terminal alkyne were investigated under the optimal conditions (2e–2m). For substrates with a methyl-substituted aryl group, the order of activity is ortho > para > meta in terms of yields (2e–2i). Interestingly, the bulky substrate also provided the desired product 2j in 61% yield. It is noted that naphthalen-1-yl-, 4-MeOC6H4- or 4-ClC6H4-susbtituted substrates are compatible with the optimal conditions (2k–2m). Gratifyingly, cyclohexenylalkyne was also suitable for the reaction (2n), which makes this methodology more useful for the preparation of pharmaceuticals and natural products. However, aliphatic alkyne was unsuitable for the reaction (2o). Two substituents, methyl or fluoro groups, on the N-aryl ring were tolerated and produced the target products 2p–2q in 56% and 28% yields, respectively. It is noteworthy that the yields of 2 were enhanced using diglyme medium (2j, 2m, 2n and 2q).
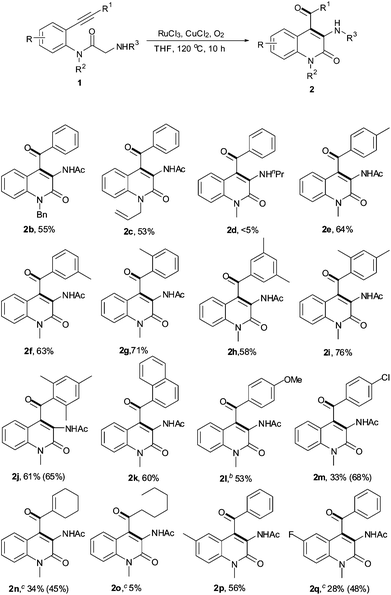
Some control experiments are summarized in Scheme 2. The treatment of substrate 1a with H218O, RuCl3, CuCl2 and O2 afforded the corresponding 18O-containing products, which were determined by GC-MS analysis,† suggesting that the oxygen atom is from water through the hydration reaction (eqn (1) in Scheme 2). Interestingly, a monodeuterated product, 3a-D1D1, was isolated from the corresponding deuterated substrate 1a-D2D2. However, about 20% D was missed in the cyclization reaction (see supplementary information†). Surprisingly, product 3a could be dehydrogenated to product 2a under the standard conditions, even in the presence of CuCl2 alone.10,11 Notably, no product 4a was observed from either 2a or 3a under the same conditions. The results in eqns (2) and (3) disclose that the dehydrogenation and isomerization between 2a and 3a took place under the optimal conditions. The value of the intermolecular kinetic isotope effect (kH/kD = 2.3) implies that the C–H bond functionalization is the rate-limiting step.10,12
 | ||
| Scheme 2 Control experiments. | ||
The reaction profiles showed that substrate 1a was consumed completely in 0.5 h. However, the yields of products 2a, 3a and 4a still changed slightly with time after 0.5 h, from a decrease to an increase for 2a, a decrease throughout the reaction for 3a, and from an increase to a decrease for 4a between 0.5–1.5 h (Fig. 1). The yield of 3a has a maximal peak at 0.5 h because some 3a is converted to 2a. The profile of 2a is contrary to that of 4a between 0.5–1.5 h, suggesting that 4a may be from the same intermediate with 2a and this intermediate is stable to some extent between 0.5–1.5 h.† Interestingly, the in situFTIR analysis also indicated that the concentration of the two peaks, 1975 cm−1 and 2128 cm−1, increased in the time period 0–1.5 h during the reaction of 1a in diglyme.†13 However, they disappeared after quenching the reaction.
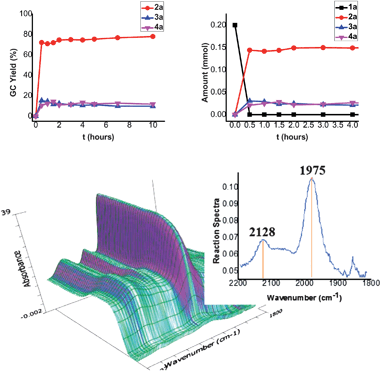 | ||
| Fig. 1 Reaction profiles (in THF) and data for the in situFTIR analysis (in diglyme) during the reaction of 1a. | ||
A possible mechanism, outlined in Scheme 3, was proposed on the basis of the present results.†7–13 The oxidation of RuCl3 produces the active RuVOCl3 species.9g,10 The complexation of RuVOCl3 with an alkyne and a nitrogen atom affords intermediate A, followed by the addition of RuVOCl3 and H2O to intermediate A, which yields intermediate B. Isomerization/C–H functionalization of intermediate B gives intermediate C10 and intermediate D.14 Reductive elimination of intermediate C results in product 3a. Intermediate D may undergo reductive elimination by two pathways:†10,13 (1) α-H elimination leading to 2a and (2) α-NHAc elimination to 4a. The conversion of 3a to 2a may proceed via a dehydrogenation of amide/isomerization process in the presence of Ru or/and Cu, which is supported by eqns (2) and (3).10,11
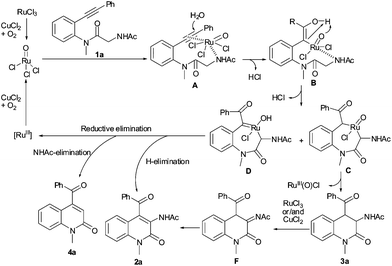 | ||
| Scheme 3 Possible mechanism. | ||
In summary, we have developed a novel ruthenium-catalyzed intramolecular carbocyclization of alkynes with an sp3carbon in an amide, including an oxidative deprotonation process. This tandem method is valuable for preparing bioactive amino-substituted quinolinones1,2 with high functional group compatibility through the oxidative hydration–deprotonation–carbocyclization tandem reaction. The mechanism was also discussed according to the 18O-labeled experiment, intermolecular kinetic isotope effect experiment and in situFTIR analysis.
This research was supported by the National Natural Science Foundation of China (No. 20872112), Fundamental Research Funds for the Central Universities (Hunan university) and Hunan Provincial Innovation Foundation For Postgraduate for financial support.
Notes and references
- For selected papers, see: (a) P. Desos, J. M. Lepagnol, P. Morain, P. Lestage and A. A. Cordi, J. Med. Chem., 1996, 39, 197 CrossRef CAS; (b) M. Patel, R. J. McHugh Jr., B. C. Cordova, R. M. Klabe, L. T. Bacheler, S. Erickson-Viitanen and J. D. Rodgers, Bioorg. Med. Chem. Lett., 2001, 11, 1943 CrossRef CAS; (c) P. R. Angibaud, G. C. Sanz, M. G. Venet, P. Muller, Janssen Pharmaceutica NV, EP 1 162 201, 2001; (d) T. Fujioka, S. Teramoto, T. Mori, T. Hosokawa, T. Sumida, M. Tominaga and Y. Yabuuchi, J. Med. Chem., 1992, 35, 3607 CrossRef CAS; (e) A. L. Hopkins, J. Ren, J. Milton, R. J. Hazen, J. H. Chan, D. I. Stuart and D. K. Stammers, J. Med. Chem., 2004, 47, 5912 CrossRef CAS; (f) B. C. Capell, M. Olive, M. R. Erdos, K. Cao, D. A. Faddah, U. L. Tavarez, K. N. Conneely, X. Qu, H. San, S. K. Ganesh, X. Chen, H. Avallone, F. D. Kolodgie, R. Virmani, E. G. Nabel and F. S. Collins, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15902 CrossRef CAS; (g) P. Cheng, Q. Zhang, Y.-B. Ma, Z.-Y. Jiang, X.-M. Zhang, F.-X. Zhang and J.-J. Chen, Bioorg. Med. Chem. Lett., 2008, 18, 3787 CrossRef CAS; (h) J. M. Kraus, C. L. M. J. Verlinde, M. Karimi, G. I. Lepesheva, M. H. Gelb and F. S. Buckner, J. Med. Chem., 2009, 52, 1639 CrossRef CAS; (i) D. S. Hong, S. M. Sebti, R. A. Newman, M. A. Blaskovich, L. Ye, R. F. Gagel, S. Moulder, J. J. Wheler, A. Naing, N. M. Tannir, C. S. Ng, S. I. Sherman, A. K. E. Naggar, R. Khan, J. Trent, J. J. Wright and R. Kurzrock, Clin. Cancer Res., 2009, 15, 7061 CrossRef CAS.
- (a) P. Friedlander, Ber. Dtsch. Chem. Ges., 1882, 15, 2572 CrossRef; (b) L. Knorr, Ber. Dtsch. Chem. Ges., 1883, 16, 2593 CrossRef; (c) G. Jones, Comprehensive Heterocyclic Chemistry II, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, New York, 1996, vol. 5, p. 167 Search PubMed.
- For papers on the synthesis of quinolinones by transition metal-catalyzed carbonylative annulation, see: (a) A. C. Tadd, A. Matsuno, M. R. Fielding and M. C. Willis, Org. Lett., 2009, 11, 583 CrossRef CAS; (b) Z. Liu, C. Shi and Y. Chen, Synlett, 2008, 1734 CAS; (c) G. Battistuzzi, R. Bernini, S. Cacchi, I. De Salve and G. Fabrizi, Adv. Synth. Catal., 2007, 349, 297 CrossRef CAS; (d) R. Bernini, S. Cacchi, G. Fabrizi and A. Sferrazza, Heterocycles, 2006, 69, 99 CrossRef CAS; (e) D. V. Kadnikov and R. C. Larock, J. Org. Chem., 2004, 69, 6772 CrossRef CAS.
- (a) C. Jia, T. Kitamura and Y. Fujiwara, Acc. Chem. Res., 2001, 34, 633 CrossRef CAS; Pd: (b) C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992 CrossRef CAS; (c) C. Jia, W. Lu, J. Oyamada, T. Kitamura, K. Matsuda, M. Irie and Y. Fujiwara, J. Am. Chem. Soc., 2000, 122, 7252 CrossRef CAS; (d) C. Jia, D. Piao, T. Kitamura and Y. Fujiwara, J. Org. Chem., 2000, 65, 7516 CrossRef CAS; (e) D.-J. Tang, B.-X. Tang and J.-H. Li, J. Org. Chem., 2009, 74, 6749 CrossRef CAS; Pt: (f) C. Nevado and A. M. Echavarren, Chem.–Eur. J., 2005, 11, 3155 CrossRef CAS; Au: (g) D. B. England and A. Padwa, Org. Lett., 2008, 10, 3631 CrossRef CAS; Ru: (h) M. Y. Yoon, J. H. Kim, D. S. Choi, U. S. Shin, J. Y. Lee and C. E. Song, Adv. Synth. Catal., 2007, 349, 1725 CrossRef CAS; Lewis acids: (i) C. E. Song, D.-U. Jung, S. Y. Choung, E. J. Roh and S.-G. Lee, Angew. Chem., Int. Ed., 2004, 43, 6183 CrossRef CAS.
- (a) L.-C. Campeau and K. Fagnou, Chem. Commun., 2006, 1253 RSC; (b) L.-C. Campeau, M. Parisien, A. Jean and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 581 CrossRef CAS.
- (a) M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 14058 CrossRef CAS; (b) K. Inamoto, T. Saito, K. Hiroya and T. Doi, J. Org. Chem., 2010, 75, 3900 CrossRef CAS; (c) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS; (d) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011 10.1039/c1cs15083a.
- (a) D. A. Culkin and J. F. Hartwig, Acc. Chem. Res., 2003, 36, 234 CrossRef CAS; (b) F. Bellina and R. Rossi, Chem. Rev., 2010, 110, 1082 CrossRef CAS; (c) F. Dénès, A. Pérez-Luna and F. Chemla, Chem. Rev., 2010, 110, 2366 CrossRef.
- For a paper on a Cu-catalyzed oxidative dehydrogenation reaction between an sp3carbon adjacent to a nitrogen atom at the α-position of an amide and an sp C–H bond of a terminal alkyne under neutral conditions, see: L. Zhao and C.-J. Li, Angew. Chem., Int. Ed., 2008, 47, 7075 CrossRef CAS.
- (a) S.-I. Murahashi, T. Naota and K. Yonemura, J. Am. Chem. Soc., 1988, 110, 8256 CrossRef CAS; (b) S.-I. Murahashi, T. Naota, T. Kuwabara, T. Saito, H. Kumobayashi and S. Akutagawa, J. Am. Chem. Soc., 1990, 112, 7820 CrossRef CAS; (c) S.-I. Murahashi, T. Naota and T. Nakato, Synlett, 1992, 835 CrossRef CAS; (d) S.-I. Murahashi, N. Komiya, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS; (e) S.-I. Murahashi, T. Nakae, H. Terai and N. Komiya, J. Am. Chem. Soc., 2008, 130, 11005 CrossRef CAS; (f) M.-Z. Wang, C.-Y. Zhou, M.-K. Wong and C.-M. Che, Chem.–Eur. J., 2010, 16, 5723 CAS; (g) J. T. Groves, M. Bonchio, T. Carofiglio and K. Shalyaev, J. Am. Chem. Soc., 1996, 118, 8961 CrossRef CAS; For reviews, see: (h) S.-I. Murahashi and N. Komiya, in Modern Oxidation Methods, ed. J.-E. Bkckvall,Wiley-VCH: Weinheim, 2004, p. 165 Search PubMed; (i) S.-I. Murahashi, Angew. Chem., Int. Ed. Engl., 1995, 34, 2443 CrossRef CAS; (j) T. Naota, H. Takaya and S.-I. Murahashi, Chem. Rev., 1998, 98, 2599 CrossRef CAS; (k) C.-J. Li, Acc. Chem. Res., 2009, 42, 335 CrossRef CAS.
- (a) K. Yamaguchi and N. Mizuno, Angew. Chem., Int. Ed., 2003, 42, 1480 CrossRef CAS; (b) Ruthenium in Organic Synthesis, ed. S.-I. Murahashi,Wiley-VCH: Weinheim, 2004 Search PubMed; (c) I. W. C. E. Arends, T. Kodama and R. A. Sheldon, in Ruthenium Catalysts in Fine Chemistry; Topics in Organometallic Chemistry, ed. C. Bruneau and P. H. Dixneuf, Springer-Verlag: Berlin, Heidelberg, 2004, vol. 11, p. 277 Search PubMed.
- CuCl2/O2 (a) M. Shimizu, H. Orita, T. Hayakawa, K. Suzuki and K. Takehira, Heterocycles, 1995, 41, 773 CrossRef CAS; Ru(OH) x/Al2O3: (b) K. Kamata, J. Kasai, K. Yamaguchi and N. Mizuno, Org. Lett., 2004, 6, 3577 CrossRef CAS.
- (a) W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS; (b) W. D. Jones and F. Feher, Acc. Chem. Res., 1989, 22, 91 CrossRef CAS.
- (a) Structure Determination of Organic Compounds, Tables of Spectral Data, 4 ed.; E. Pretsch, P. Bühlmannn, C. Affolter, ed.; Springer-Verlag: Berlin Heidelberg, 2009, p. 269 Search PubMed; (b) Y. Hu, J. Liu, Z. Lü, X. Luo, H. Zhang, Y. Lan and A. Lei, J. Am. Chem. Soc., 2010, 132, 3153 CrossRef CAS; (c) Q. Liu, Y. Lan, J. Liu, G. Li, Y.-D. Wu and A. Lei, J. Am. Chem. Soc., 2009, 131, 10201 CrossRef CAS.
- B. M. Trost and M. T. Rudd, J. Am. Chem. Soc., 2002, 124, 4178 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Table S1, Figs S1(GC-MS analysis), S2 (FTIR analysis) and S3 (the reaction profiles). CCDC reference numbers 832462. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1sc00423a |
| ‡ These authors contributed equally |
| This journal is © The Royal Society of Chemistry 2011 |