A rapid method for detection of genetically modified organisms based on magnetic separation and surface-enhanced Raman scattering†
Burcu
Guven
a,
İsmail Hakkı
Boyacı
*a,
Ugur
Tamer
b and
Pınar
Çalık
c
aDepartment of Food Engineering, Faculty of Engineering, Hacettepe University, Beytepe, Ankara, 06800, Turkey. E-mail: ihb@hacettepe.edu.tr; Fax: +90 312 299 21 23; Tel: +90 312 297 61 46
bDepartment of Analytical Chemistry, Faculty of Pharmacy, Gazi University, 06330, Ankara, Turkey
cDepartment of Chemical Engineering, Industrial Biotechnology and Metabolic Engineering Laboratory, Middle East Technical University, 06531, Ankara, Turkey
First published on 3rd November 2011
Abstract
In this study, a new method combining magnetic separation (MS) and surface-enhanced Raman scattering (SERS) was developed to detect genetically modified organisms (GMOs). An oligonucleotide probe which is specific for 35 S DNA target was immobilized onto gold coated magnetic nanospheres to form oligonucleotide-coated nanoparticles. A self assembled monolayer was formed on gold nanorods using 5,5′-dithiobis (2-nitrobenzoic acid) (DTNB) and the second probe of the 35 S DNA target was immobilized on the activated nanorod surfaces. Probes on the nanoparticles were hybridized with the target oligonucleotide. Optimization parameters for hybridization were investigated by high performance liquid chromatography. Optimum hybridization parameters were determined as: 4 μM probe concentration, 20 min immobilization time, 30 min hybridization time, 55 °C hybridization temperature, 750 mM buffer salt concentration and pH: 7.4. Quantification of the target concentration was performed via SERS spectra of DTNB on the nanorods. The correlation between the target concentration and the SERS signal was found to be linear within the range of 25–100 nM. The analyses were performed with only one hybridization step in 40 min. Real sample analysis was conducted using Bt-176 maize sample. The results showed that the developed MS-SERS assay is capable of detecting GMOs in a rapid and selective manner.
1. Introduction
A genetically modified organism (GMO) is usually defined as a living organism whose genetic composition has been altered by gene technology. This modification involves DNA isolation, DNA modification, and transfer of DNA into the target organism genome. A GMO is composed of at least three elements: (1) the promoter element, which functions as a start signal for reading of the inserted/altered gene; (2) the gene that has been inserted/altered, which is coding for a specific selected feature; (3) the terminator element, which functions as a stop signal for reading of the inserted/altered gene. Also several other elements are used for controlling and stabilizing the gene function.1 The most common elements in GMO structures, the 35 S promoter from Cauliflower mosaic virus (p35 S) and the terminator from napoline synthetase gene of Agrobacterium tumefaciens (tNOS), have been typical targets and used for screening the presence of GMOs.2 Gene modification is expected to improve both breeding technology and the development of new plant varieties of high quality and yield, with characteristics such as pest and disease resistance and environmental stress tolerance, etc.3Detection and identification of GMOs are gaining worldwide attention due to the expansion of the GMOs. The detection and identification of GMOs have been generally achieved with DNA- and protein-based methods. Protein-based methods are unable to detect a genetic modification if the modified gene is inactive in the cells.1 DNA-based methods for detecting GMOs are based on the complementaries of two strands of DNA double helix that hybridize in a sequence-specific manner.4
Since its discovery, SERS has received more and more attention from researchers around the world,5,6 not only because of its high sensitivity, the small volume of sample needed,7 and the possible wide applicability8,9 but also due to the stable and specific signal.10 The SERS detection of various biomolecules or species has been reported, such as proteins, enzymes, viruses, bacteria, cancer markers, and nucleotides.11 DNA can show well-distinguished SERS bands while the sugar or phosphate groups on the backbone have little interference. However, the direct detection of DNA sequence at low concentration is difficult with SERS.12 The resulting spectrum strongly depends on the sequence of the bases,13 and also on the binding modes of DNA strands,14 which results in difficulty to identify the DNA sequence. Additionally, the methodologies used for DNA recognition are usually based on the hybridization of the target nucleic acid with its complementary bases, as there is a little difference between their Raman spectra.11 Therefore, reliable and reproducible SERS active substrates are needed to label the DNA15 such as cresyl fast violet,16 Rhodamine B,17 Rhodamine 6G and others.18 Some metal nanoparticles have also been conjugated with DNA and used to enhance the Raman intensity.19,20
Nanoparticle-based Raman tags have also advantages in comparison with other nanoparticle tags. Nanoparticle-based Raman tags have been developed by some research groups.21–24 There are two kinds of nanoparticle-based Raman tags reported in the literature, one of which is prepared by the direct attachment of both a Raman reporter and biomolecule to nanoparticle; whereas the other is prepared by the dye-embedded core-shell nanoparticle-based Raman tag.25
Magnetic nanoparticles are among the most frequently used as Raman tags. Magnetic nanoparticles have become interesting tools for biological molecules and cells. They have been used for concentration, separation, purification and identification of molecules and cells26 besides immobilization of proteins and enzymes, bioseparation, assay, drug delivery and biosensors. The magnetic nanoparticles enable the isolation or extraction of a target molecule or substance by the use of an external magnetic field. The usage of magnetic nanoparticles not only possesses the advantage of superparamagnetism, but also preconcentrates the analyte in order to amplify the Raman signal efficiently.27 In this study, we have demonstrated that the magnetic nanoparticles could can be used for developing a reliable and selective SERS based homogeneous sandwich assay. The DNA probes were immobilized on spherical magnetic gold nanoparticles and rod shaped gold nanoparticles and hybridized with the target oligonucleotide. Rod shaped nanoparticles were used as SERS reporters. Then the SERS signals were obtained and the calibration curve was plotted to measure the different concentrations of target oligonucleotide. Optimum hybridization parameters were determined by using high performance liquid chromatography. The selectivity and specificity tests of the developed assay were examined with positive and negative controls. Finally, this assay was applied to the 35 S sequence of Bt-176 maize sample. The analytical performance of the SERS based sandwich assay system with respect to linear range, detection limit, response time, and selectivity is presented and discussed.
2. Experimental
2.1. Reagents and materials
Hydrogen tetrachloroaurate (HAuCl4), hexadecyltrimethyl-ammoniumbromide (CTAB), L-ascorbic acid (AA) were obtained from Sigma–Aldrich (Taufkirchen, Germany). 11-mercaptoundecanoic acid (11-MUA), 98% ethanolamine, N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), FeCl3, 2-morpholino ethanesulfonic acid monohydrate (MES) were obtained from Sigma-Aldrich (Steinheim, Germany). Silver nitrate (AgNO3), sodium borohydride (NaBH4), di-sodium hydrogen phosphate (Na2HPO4) solution (30%), FeSO4·7H2O, perchloric acid (HClO4) and absolute ethanol were purchased from Merck (Darmstadt, Germany). 5,5-Dithiobis (2-nitrobenzoic acid) (DTNB) was obtained from Acros (Morris Plains, NJ, USA). N-Hydroxysulfosuccinimide sodium salt (NHS) is the product of Pierce Biotechnology (Bonn, Germany). Tween 20 was obtained from AppliChem, Biochemica (Darmstadt, Germany). NaCl, Na2HPO4, and KH2PO4 were obtained from J.T. Baker (Deventer, The Netherlands) and used as phosphate buffered saline (PBS). All reagents were used as received.PCR buffer, MgCl2, dNTPs, Taq DNA polymerase and forward primer were obtained from Fermentas Inc. (Vilnius, Lithuania) and oligonucleotides were purchased from Alpha DNA (Quebec, Canada). The sequences are as shown in Table 1.
| Probe 1 | 5′-NH2-AAA AAT CGG CAG AGG CAT-3′ |
| Probe 2 | 5′-CGA TGG CCT TTC CAA AAA-NH2 -3′ |
| Target | 5′-GGA AAG GCC ATC GTT GAA GAT GCC TCT GCC GA-3′ |
| Nonsense Sequence | 5′-CAT TTT GGA CAA AGC GTC TAC GCT GCA G-3′ |
| Forward Primer p 35 S-cf3 | 5′-CCACGTCTTCAAAGCAAGTGG-3′ |
| Reverse Primer | 5′-NH2-AAAAATCGGCAGAGGCAT-3′ |
After and before the NH2 groups, five adenine bases of DNA probes were added as spacer parts. Bt-176 maize DNA template was obtained from Central Laboratory of Middle East Technical University (Ankara, Turkey).
2.2. Solutions and buffers
PBS (20 mM, pH 7.4) was prepared by using Na2HPO4, KH2PO4, NaCl, and adjusting the pH with HCl or NaOH. PBS was prepared with 150 mM NaCl concentration as washing buffer and 750 mM NaCl concentration as hybridization buffer. PBST was prepared mixing PBS with 0.05% Tween 20 (v/v). 2-Morpholino-ethanesulfonic acid monohydrate buffer (MES; 0.05 M, pH 6.5) was prepared by adjusting the pH with NaOH. EDC/NHS solution (0.2 M EDC and 0.05 M NHS) was prepared with 0.05 M MES buffer. All solutions were prepared with Milli-Q quality water (18 MΩ cm) to reach the desired concentrations. The buffers involved in this work are as follows: DNA immobilization buffer, 50 mM MES buffer (pH 6.5); buffer for hybridization, 20 mM phosphate buffer plus 750 mM NaCl (pH 7.4).2.3. Instrumentation
DeltaNu Examiner Raman Microscopy system (Deltanu Inc., Laramie, WY) with a 785 nm laser source, a motorized microscope stage sample holder, and a cooled charge-coupled device (CCD, at 0 °C) detector were used to detect target oligonucleotides. The instrument parameters were as follows: 20x objective, 30 μm laser spot size, 150 mW laser power, and 50 s acquisition time. Baseline correction was performed for all of the measurements. Probe concentration, immobilization time, hybridization time, hybridization temperature, buffer salt concentration, and buffer pH were optimized by using an Agilent 1100 series high performance liquid chromatography (HPLC) system integrated with a binary pump, degasser, well-plate autosampler, thermostatted column compartment, diode array detector and a personal computer with a software package (Chemstation Software) for system control and data acquisition (Agilent Technologies, Santa Clara, CA). The flow rate was set at 0.6 mL min−1 and PBS (0.1 M, pH 7.4) was used as the mobile phase. The absorbance of oligonucleotide was measured at 260 nm. PCR products were obtained from GeneAmp-9700 (Applied Biosystems, East Lyme, CT) and were used to amplify 333-bp sequence in the 35 S region of the Bt-176 maize genome.PCR products were analyzed using a gel documentation system by UVP BioImaging System (UVP, Inc., Upland, CA) and pre-cast gels were stained with ethidium bromide. The quantity of products was analyzed using a micro volume spectrophotometer (NanoDrop, Wilmington, DE).
2.4. Fabrication of nanoparticles
The fabrication of gold coated magnetic nanospheres28 and the fabrication of gold nanorods were performed by using a seed-mediated growth technique.292.5. Preparation of Raman labels
Gold nanorods were used as a Raman label by assembling DTNB to form free carboxyl groups on the surface of the nanoparticle. The self-assembled monolayer (SAM) was formed on the gold nanorod by using 50 mM DTNB for 18 h at room temperature. Then, the gold nanorods were washed with 0.05 M MES buffer and collected by the effect of centrifuge (7000 rpm, 5 min).2.6. Preparation of probe coated nanoparticles and sandwich assay design
The schematic illustration of the stepwise assay design process of the nanoparticle preparation, immobilization, hybridization and selective detection of the target is shown in Fig. 1.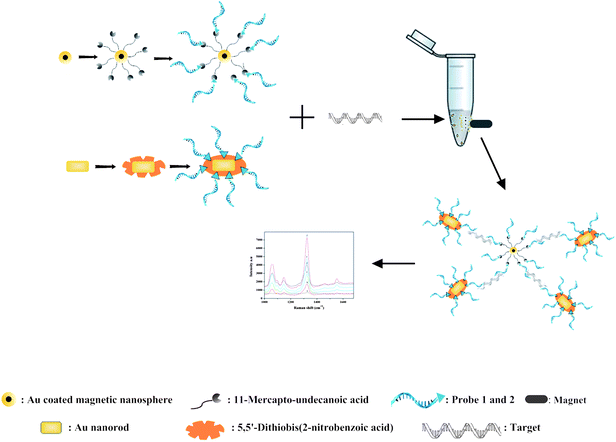 | ||
| Fig. 1 Schematic illustration of MS-SERS based sandwich assay for the target oligonucleotide. | ||
The surfaces of the gold coated magnetic nanospheres were modified with 150 mM 11-MUA in absolute ethanol overnight to form the SAM. The nanoparticles were collected by using a permanent magnet and washed with 0.05 M MES (pH 6.5) buffer. For surface activation over the carboxyl groups, the nanoparticles were treated with 1 mL of EDC/NHS solution for 40 min. The nanoparticles were separated magnetically and were washed twice with 0.05 M MES buffer solution. After this procedure, the nanoparticle surfaces were immobilized with DNA probes by incubating for 20 min to form a covalent bond between the amino group of the probe and the carboxyl group of the nanoparticles. The nanoparticles were collected with a magnet and washed twice with MES buffer. For avoiding non-specific interaction 1% (v/v) ethanolamine was used for blocking active groups on the surface of the nanoparticles.
The surfaces of the gold nanorods were modified with 50 mM DTNB in absolute ethanol overnight to form a SAM. The nanoparticles were collected by centrifuge and washed with 0.05 M MES buffer. Similar with the magnetic nanoparticles, surface activation of the gold nanorods, probe immobilization and then blocking of the particle surfaces were performed. After each step, the washing procedure was applied twice by MES buffer.
At the hybridization step, the probe coated magnetic nanospheres and probe coated nanorods were mixed, the mixed solution was centrifuged to separate the liquid phase and then the target oligonucleotide solution was added to the mixed nanoparticle-probe complex. The hybridization step was performed at 55 °C in PBS solution for 30 min to form a probe-target conjugation. Then, the washing procedure was performed with PBS and PBST buffers. In all washing steps, nanoparticles were dispersed in liquid media using an ultrasonic bath.
2.7. Optimization of the parameters
The probe (Fig. S1†) and target (Fig. S3†) oligonucleotide solutions were optimized using HPLC and calibrated with increased concentrations (Fig. S2 and S4†). Then the immobilization and hybridization steps were performed as mentioned above and free probe or target oligonucleotides were separated using a magnet. The quantity of free probe and target oligonucleotides in the solution were determined using HPLC. The relative capturing yields were calculated using the quantity of free and noninteracted probe and target oligonucleotides (Fig. S5†). Quantitative HPLC analysis was performed using a gel filtration column (Zorbax 4.6 × 250 mm, 5 mm GF-250, Agilent Technologies, CA) and an aqueous mobile phase containing 0.1 M PBS (pH 7.4) with a flow rate of 0.6 mL min−1. The injected volume was 10 μL at 25 °C and the detector was set at 260 nm. The retention time was approximately 15 min for the probe and target oligonucleotide solutions, under certain conditions. Experiments were performed with triplicates for each data point and average and standard deviations were calculated using Microsoft Excel®.The immobilization and hybridization parameters such as probe concentration, immobilization time, hybridization time, hybridization temperature, buffer salt concentration and pH were optimized by using HPLC. The probe oligonucleotide was immobilized on the gold coated magnetic nanospheres. The effects of the probe concentration and probe-nanoparticle immobilization time were determined. The gold coated magnetic nanospheres were mixed with the probe solutions at concentrations between 2 and 10 μM. Nanoparticles were mixed with the probe solutions at immobilization times between 1 and 45 min. The target-probe complexes were hybridized between 10 and 60 min. Furthermore, the target-probe complexes were hybridized between 25 and 95 °C. The buffer salt concentration and pH parameters were also examined in the range of 150–1500 mM and 6.5–8.2, respectively. In order to eliminate non-specific interaction, the washing procedure was modified after hybridization. The modified washing procedure was performed 3 times with PBS, 3 times with PBST, and 3 times with PBS, respectively.
2.8. Detection of target oligonucleotide using MS-SERS
DTNB labeled gold nanorod-probe 1 and gold coated magnetic nanosphere-probe 2 complexes were hybridized with target oligonucleotide at 55 °C for 30 min to form a sandwich structure in the solution. Then sandwich complex was separated magnetically from the solution and the washing procedure was repeated. Just after the washing procedure, the complex was re-suspended in 100 μL PBS. 5 μL of suspension was placed on the thin layer chromatography (TLC) paper. The integration time was 50 s and all experiments were performed three times. The average value of three SERS spectra for DTNB was obtained. The Raman spectra corresponding to each concentration of the target were recorded. The band intensity at 1370 cm−1versus the target concentration calibration curve was plotted. The linearity range and coefficient of determination (R2), LOD and LOQ values were calculated from the calibration curves.30The specificity of the developed assay was evaluated by negative and positive control assays. The negative control was performed by interaction of probe 1 and probe 2 without target oligonucleotide sequence. Two positive controls were done with the target oligonucleotide sequence. There were interactions of probe 1 and target (without probe 2) and probe 2 and target (without probe 1). The selectivity of the assay was examined with a nonsense sequence. The accuracy of the developed method was investigated with the Bt-176 maize genome which was isolated by the Gene Amp 2400 method.31 The forward primer p 35 S-cf3 and the reverse primer probe 1 of the 35 S regions were used. PCR amplification was carried out in 50 μL 1x PCR buffer (pH 8.3). The contents of the PCR buffer were MgCl2 (2.5 mM), dNTPs (0.2 mM), forward and reverse primers (0.5 μM), Taq DNA polymerase (0.025 U/μL) and Bt-176 maize DNA template (2 μL % 0.5). Amplification was achieved with an initial denaturation step at 95 °C for 4 min followed by a 40 cycle process that includes a denaturation step for 1 min at 95 °C, an annealing step for 1 min at 62 °C, an extension step for 1 min at 72 °C, and a final extension at 72 °C for 10 min. Dilution of the PCR product was prepared. The amount of diluted PCR product was analyzed using the developed MS-SERS assay. The PCR product was denatured at 94–96 °C to obtain a single strand in the presence of the probe immobilized gold coated magnetic nanospheres and gold nanorods. Hybridization was performed at 55 °C for 30 min, then the washing procedure was applied and the SERS measurement was performed. The target sequence concentration was calculated using the SERS intensity and calibration graph. They were also quantified using a micro volume spectrophotometer and the results of the methods were compared.
Experiments were performed in triplicate, average and standard deviations were calculated, and data analysis was performed using Microsoft Excel®.
3. Results and discussion
3.1. Fabrication of the nanoparticles
Gold coated magnetic nanospheres and gold nanorods were produced to perform magnetic separation and preparation of the SERS label, respectively. The gold coated magnetic nanospheres have average diameter of 12.5 ± 3 nm. The length and width of the synthesized gold nanorods were determined as 45 nm and 15 nm, respectively. Although the gold coated nanospheres have a plasmon band at 521 nm, the plasmon bands of the gold nanorods were determined for the transverse plasmon and the longitudinal plasmon as 524 nm and 665 nm, respectively. In this study, DTNB labeled nanorods as the SERS reporter were used due to generating a strong Raman signal based on the research of Temur and coworkers.29,32 The geometrical shapes of the nanoparticles affect the SERS intensity. It is also reported that sharp shaped nanoparticles enhance SERS intensity.33 The characterization data of the nanoparticles were published in our previous articles.28,29,34Non-spherical gold nanorod particles act as electromagnetic hot spots for SERS owing to the facile tunability of the plasmonic properties and presence of sharp corners and edges. Such anisotropic nanoparticles display strong polarization dependence in their plasmonic properties, exhibiting significantly higher SERS intensity with certain orientations. In addition, the degree of surface plasmon coupling increases at the gaps or junctions of nanoparticles with the creation of hot spots. Based on these considerations, creating hot spots using magnetic nanoparticle-nanorod interactions results in high sensitivity and selectivity of the developed systems.35
3.2. Optimization of capturing yield
The success of the developed assay is directly related with the efficiencies of the analysis steps. On the basis of this knowledge, the optimum values for capturing of target molecule on the nanoparticle surfaces were determined in the first part of the study. The effects of probe concentration, immobilization time, hybridization time, hybridization temperature, buffer salt concentration and buffer pH on the target molecule capturing yield were investigated and the results are given in Fig. 2. Optimum values for the investigated parameters were determined as: probe concentration (Fig. 2a): 4 μM which is equivalent to 0.001 μmol probe/μg NP, probe immobilization time (Fig. 2b): 20 min, hybridization time (Fig. 2c): 30 min, hybridization temperature (Fig. 2d): 55 °C, buffer salt concentration (Fig. 2e): 750 mM and buffer pH (Fig. 2f): 7.4. Low relative capturing yields were observed during the lower and higher values of these parameters rather than the optimum values. The optimum parameter values were used in the development of the MS-SERS assay for detection of target oligonucleotides. To avoid non-specific interaction, the washing procedure which was developed in our previous study was applied.34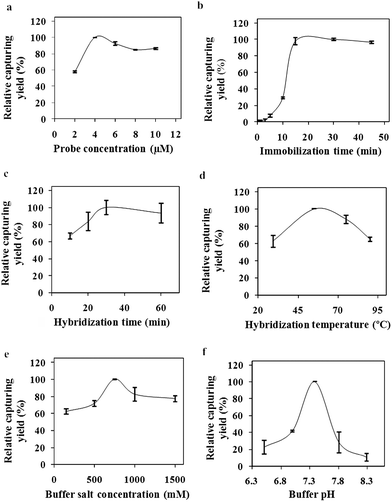 | ||
| Fig. 2 Optimization parameters (a) immobilization concentration, (b) immobilization time, (c) hybridization time, (d) hybridization temperature, (e) buffer salt concentration, (f) buffer salt pH. | ||
As it can be seen from the published studies, the immobilization times change between 15 min36 to 30 min.37 The immobilization time of the developed method was shown to be one of the shortest methods. The hybridization time for the nanoparticle based electrochemical DNA analysis method, absorbance based, SERS based and fluorescence based methods were found as 20 min,38 40 min,39 60 min25 and 120 min,40 respectively. The hybridization temperature was chosen as 55 °C in most studies.39,41 Tsuruoka and Karube42 showed that high salt concentrations have accelerated the hybridization process. The optimum buffer salt concentration was found as 750 mM in our study. Hybridization buffer salt concentrations were given as 600 mM25 and 1000 mM43 in published studies. Also the hybridization buffer pH was chosen as 725,44 and 7.4.43
3.3. Target detection using MS-SERS
The SERS spectra for target assays conducted using a Raman label constructed from a gold nanorod are shown in Fig. 3. which displays a typical SERS response of the assay system after the addition of various concentrations of the target oligonucleotide.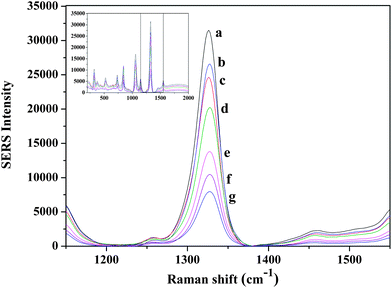 | ||
| Fig. 3 Symmetric NO2 stretching bands of DTNB range from 1000 nM to 0 nM target concentration obtained with rod shaped nanoparticles (a) 1000 nM, (b) 500 nM, (c) 100 nM, (d) 75 nM, (e) 50 nM, (f) 25 nM (g) 0 nM target concentration. | ||
The spectra contain features which are attributable to the Raman label (reporter molecule) and are dominated by bands representative of the DTNB-based adlayer (e.g., the symmetric nitro stretch (Vs (NO2)) at 1370 cm−1. The detection is based on the characteristic feature of the Raman tag and then quantified by its intensity. The calibration curve was plotted with the changes of the band intensities of DTNB vs. the different concentrations of target oligonucleotide (0–1000 nM). As shown in Fig. 4, the intensities of the Raman spectra were obtained in the presence of different concentrations of target oligonucleotide. A good linear correlation (R2 = 0.998) was obtained within the range of 25–100 nM target oligonucleotide concentration. The sensitivity of the developed assay was investigated and the LOD and LOQ of the proposed method for the target were found to be 11 nM and 34 nM, respectively.
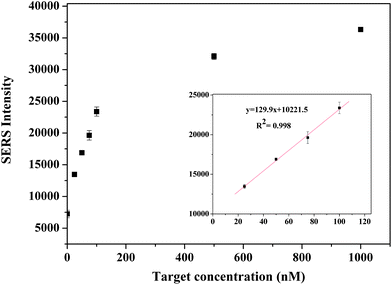 | ||
| Fig. 4 Calibration curve for the target oligonucleotide in the range of 0–1000 nM. | ||
The specificity of the developed assay was evaluated and the band intensities of the negative and positive controls were obtained. The selectivity test was performed using the nonsense sequence. Intensities obtained from the specificity and selectivity tests were compared with the intensities of the LOD and LOQ concentrations (Fig. 5). Shown in Fig. 5, the band intensities of the specificity and selectivity test data were lower than the intensity of the LOD value. It means that the developed MS-SERS assay is reliable and selective to only the target oligonucleotide. When the band intensities of the target oligonucleotide (Fig. 3) were compared with the specificity and selectivity data (Fig. 5), the intensities that belong to the target oligonucleotide were found to be much greater.
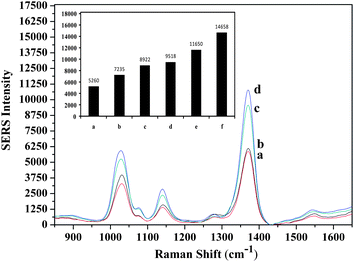 | ||
| Fig. 5 Evaluation of the specificity and selectivity of the developed method (a) without probe 1, (b) without target, (c) without probe 2, (d) nonsense sequence, (e) intensity at LOD concentration, (f) intensity at LOQ concentration. | ||
The amplification of the Bt-176 maize sample was performed with PCR. In order to observe the variation of intensity, different concentrations of PCR product were obtained and analyzed with the developed assay and also with NanoDrop. The concentrations were found to be 27.0 ± 0.21 nM and 81.0 ± 0.43 nM by NanoDrop. The concentrations measured by using the developed MS-SERS assay are 28.2 ± 4.3 nM and 91.8 ± 5.4 nM. As a comparison, similar results were obtained with NanoDrop and the proposed assay system. As a consequence, it is said that the proposed method could be applied in real samples.
The working range of the developed assay is between 25 and 100 nM with a detection limit of 11 nM and the assay time is less than 40 min including the hybridization procedure (30 min) and SERS measurement (less than 10 min). The detection limit in SERS based methods varies between 10 nM,45 100 nM43 and 10 pM,46 20 fM.18
As a comparison, it is shown that the hybridization time of the developed assay is quite short. The hybridization procedure was performed with different reaction times such as 2–3 h at room temperature47 40 min,48 60 min,49 1 h at 35 °C50 and microarray technology requires a long hybridization time (about 1–2 h) due to the diffusion-limited hybridization kinetics.51,52 Magnetic separation presents a time saving replacement and allows the flexibility of applying different end point detection methods. Furthermore, nano-sized magnetic particles have advantages due to their large surface area and nanorods can be produced at any desired size and shape.34 The reason of using gold nanorods as Raman labels instead of nanospheres is that the Raman intensity of nanorods is higher than nanospheres. It is thought that using nanorods would increase the sensitivity of developed method.29
Moreover, it was demonstrated that the method can be easily used for the determination of PCR product quantitatively. The developed assay could be advantageous over other methods because the PCR product used in this assay is double strand DNA, and it is necessary to convert it into single strand for denaturation before detection. In addition, the denaturation temperature is around 95 °C and this temperature could cause an undesirable change on the magnetic and SERS labeled nanoparticles. Although there are so many works published on related developments of DNA assays, the effects of high temperature on the analysis performance are not mentioned so far.25,53 The functional stability of the nanoparticles at high temperature could give us an opportunity to perform the assay with only one hybridization step. This opportunity would reduce the analysis time and washing procedure. A sandwich assay was performed to investigate DNA interactions and it is observed that SERS spectra were not influenced by high temperatures which are required for the denaturation step of double strand DNA. The most important point of the developed method is that the hybridization process was perfomed in one step, and the real sample was detected in a short analysis time.
4. Conclusion
In this study, we developed a rapid and selective assay for enumeration of the target oligonucleotide in Bt-176 maize samples using magnetic separation and SERS. The developed MS-SERS assay shows that magnetic separation and SERS can be used for the determination of target oligonucleotides. In this study we have also focused on the part of the 35 S DNA for screening the presence of GMOs. In studies with real samples, it was observed that the SERS intensities of the nanoparticles were not changed at denaturation temperature (94–96 °C) of double strand DNA. Therefore, all studies were performed in one hybridization step. It was shown that the developed MS-SERS assay provided rapid detection. The target oligonucleotide was detected in a real matrix in a rapid, reliable and selective manner. The sensitivity enhancement of the developed assay will be investigated in the future. Thus, rapid detection, based on molecular biology and assay techniques and/or a combination of both, has diverted attention from the time consuming enrichment steps which are needed to increase and select the target numbers for detection. It is demonstrated that the developed assay can be used for selective detection of the target oligonucleotide in 35 S sequence of Bt-176 maize samples.Acknowledgements
The authors would like to thank Dr Remziye Yilmaz, Central Laboratory Middle East Technical University, for the genomic DNA isolation from Bt-176 maize and for the technical support provided. The authors are grateful for the financial supports provided by The Scientific and Technological Research Council of Turkey; Project Number: 110T584 and 111T096.References
- M. Miraglia, K. G. Berdal, C. Brera, P. Corbisier, A. Holst-Jensen, E. J. Kok, H. J. P. Marvin, H. Schimmel, J. Rentsch, J. P. P. F. van Rie and J. Zagon, Food Chem. Toxicol., 2004, 42, 1157–1180 CrossRef CAS.
- M. Querci, M. Van den Bulcke, J. Zel, G. Van den Eede and H. Broll, Anal. Bioanal. Chem., 2010, 396, 1991–2002 CrossRef CAS.
- P. Cardarelli, M. R. Branquinho, R. T. B. Ferreira, F. P. da Cruz and A. L. Gemal, Food Control, 2005, 16, 859–866 CrossRef.
- F. E. Ahmed, Trends Biotechnol., 2002, 20, 215–223 CrossRef CAS.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Current Science, 1999, 77, 915–924 CAS.
- C. L. Haynes, A. D. McFarland and R. P. Van Duyne, Anal. Chem., 2005, 77, 338a–346a CrossRef CAS.
- P. Etchegoin, R. C. Maher, L. F. Cohen, H. Hartigan, R. J. C. Brown, M. J. T. Milton and J. C. Gallop, Chem. Phys. Lett., 2003, 375, 84–90 CrossRef CAS.
- P. Vandenabeele, H. G. M. Edwards and L. Moens, Chem. Rev., 2007, 107, 675–686 CrossRef CAS.
- M. Seydack, Biosens. Bioelectron., 2005, 20, 2454–2469 CrossRef CAS.
- M. H. Harpster, H. Zhang, A. K. Sankara-Warrier, B. H. Ray, T. R. Ward, J. P. Kollmar, K. T. Carron, J. O. Mecham, R. C. Corcoran, W. C. Wilson and P. A. Johnson, Biosens. Bioelectron., 2009, 25, 674–681 CrossRef CAS.
- C. Fang, A. Agarwal, K. D. Buddharaju, N. M. Khalid, S. M. Salim, E. Widjaja, M. V. Garland, N. Balasubramanian and D. L. Kwong, Biosens. Bioelectron., 2008, 24, 216–221 CrossRef CAS.
- C. Y. Wu, W. Y. Lo, C. R. Chiu and T. S. Yang, J. Raman Spectrosc., 2006, 37, 799–807 CrossRef CAS.
- H. Deng, V. A. Bloomfield, J. M. Benevides and G. J. Thomas, Biopolymers, 1999, 50, 656–666 CrossRef CAS.
- L. Gearheart, K. K. Caswell and C. J. Murphy, J. Biomed. Opt., 2001, 6, 111–115 CrossRef CAS.
- C. Peng, Y. Song, G. Wei, W. Zhang, Z. Li and W. F. Dong, J. Colloid Interface Sci., 2008, 317, 183–190 CrossRef CAS.
- T. Vo-Dinh, F. Yan and M. B. Wabuyele, J. Raman Spectrosc., 2005, 36, 640–647 CrossRef CAS.
- M. Culha, D. Stokes, L. R. Allain and T. Vo-Dinh, Anal. Chem., 2003, 75, 6196–6201 CrossRef CAS.
- Y. W. C. Cao, R. C. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS.
- M. B. Wabuyele and T. Vo-Dinh, Anal. Chem., 2005, 77, 7810–7815 CrossRef CAS.
- A. J. Bonham, G. Braun, I. Pavel, M. Moskovits and N. O. Reich, J. Am. Chem. Soc., 2007, 129, 14572 CrossRef CAS.
- M. H. Cho, B. H. Jun, M. S. Noh, J. Kim, G. Kim, H. Kang, M. S. Kim, Y. T. Seo, J. Baek, J. H. Kim, J. Park, S. Kim, Y. K. Kim, T. Hyeon, D. H. Jeong and Y. S. Lee, Small, 2010, 6, 119–125 CrossRef.
- K. Kim, H. J. Jang and K. S. Shin, Analyst, 2009, 134, 308–313 RSC.
- C. J. Zhong, H. Y. Park, M. J. Schadt, L. Wang, I. I. S. Lim, P. N. Njoki, S. H. Kim, M. Y. Jang and J. Luo, Langmuir, 2007, 23, 9050–9056 CrossRef.
- R. A. Alvarez-Puebla, M. Spuch-Calvar, L. Rodriguez-Lorenzo, M. P. Morales and L. M. Liz-Marzan, J. Phys. Chem. C, 2009, 113, 3373–3377 Search PubMed.
- Y. Liang, J. L. Gong, Y. Huang, Y. Zheng, J. H. Jiang, G. L. Shen and R. Q. Yu, Talanta, 2007, 72, 443–449 CrossRef CAS.
- N. Sanvicens, C. Pastells, N. Pascual and M. P. Marco, TrAC, Trends Anal. Chem., 2009, 28, 1243–1252 CrossRef CAS.
- U. Tamer, İ. H. Boyacı, E. Temur, A. Zengin, İ. Dincer and Y. Elerman, J. Nanopart. Res., 2011, 13, 3167–3176 CrossRef CAS.
- U. Tamer, Y. Gundogdu, I. H. Boyaci and K. Pekmez, J. Nanopart. Res., 2010, 12, 1187–1196 CrossRef CAS.
- E. Temur, I. H. Boyaci, U. Tamer, H. Unsal and N. Aydogan, Anal. Bioanal. Chem., 2010, 397, 1595–1604 CrossRef CAS.
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, A712 CrossRef.
- M. Lipp, P. Brodmann, K. Pietsch, J. Pauwels and E. Anklam, Journal of Aoac International, 1999, 82, 923–928 CAS.
- N. N. Yazgan, I. H. Boyaci, E. Temur, U. Tamer and A. Topcu, Talanta, 2010, 82, 631–639 CrossRef CAS.
- M. D. Porter, R. Narayanan and R. J. Lipert, Anal. Chem., 2008, 80, 2265–2271 CrossRef.
- B. Guven, N. Basaran-Akgul, E. Temur, U. Tamer and I. H. Boyaci, Analyst, 2011 Search PubMed.
- J. Irudayaraj, Y. L. Wang and K. Lee, Chem. Commun., 2010, 46, 613–615 RSC.
- A. N. Kawde and J. Wang, Electroanalysis, 2004, 16, 101–107 CrossRef.
- J. M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS.
- J. Wang, D. K. Xu, A. N. Kawde and R. Polsky, Anal. Chem., 2001, 73, 5576–5581 CrossRef CAS.
- H. G. Park, H. J. Parab, C. Jung and J. H. Lee, Biosens. Bioelectron., 2010, 26, 667–673 CrossRef.
- C. Z. Li, K. B. Male, S. Hrapovic and J. H. T. Luong, Chem. Commun., 2005, 3924–3926 RSC.
- B. J. Conner, A. A. Reyes, C. Morin, K. Itakura, R. L. Teplitz and R. B. Wallace, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 278–282 CrossRef CAS.
- M. Tsuruoka and I. Karube, Combinatorial Chemistry & High Throughput Screening, 2003, 6, 225–234 CAS.
- J. Irudayaraj, L. Sun and C. X. Yu, Anal. Chem., 2007, 79, 3981–3988 CrossRef.
- M. L. Sauthier, R. L. Carroll, C. B. Gorman and S. Franzen, Langmuir, 2002, 18, 1825–1830 CrossRef CAS.
- S. E. J. Bell and N. M. S. Sirimuthu, J. Am. Chem. Soc., 2006, 128, 15580–15581 CrossRef CAS.
- L. He, M. D. Musick, S. R. Nicewarner, F. G. Salinas, S. J. Benkovic, M. J. Natan and C. D. Keating, J. Am. Chem. Soc., 2000, 122, 9071–9077 CrossRef CAS.
- T. Vo-Dinh, H. N. Wang and J. Scaffidi, J. Biophotonics, 2010, 3, 89–102 CrossRef CAS.
- P. Du, H. X. Li, Z. H. Mei and S. F. Liu, Bioelectrochemistry, 2009, 75, 37–43 CrossRef CAS.
- Z. Q. Gao, A. Agarwal, A. D. Trigg, N. Singh, C. Fang, C. H. Tung, Y. Fan, K. D. Buddharaju and J. M. Kong, Anal. Chem., 2007, 79, 3291–3297 CrossRef CAS.
- J. Wang, S. Zhang and Y. Zhang, Anal Chem, 2010, 396, 304–309 CAS.
- A. W. Peterson, R. J. Heaton and R. Georgiadis, J. Am. Chem. Soc., 2000, 122, 7837–7838 CrossRef CAS.
- M. Jobs, S. Fredriksson, A. J. Brookes and U. Landegren, Anal. Chem., 2002, 74, 199–202 CrossRef CAS.
- H. Zhang, M. H. Harpster, H. J. Park and P. A. Johnson, Anal. Chem., 2011, 83, 254–260 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1an15629b |
| This journal is © The Royal Society of Chemistry 2012 |