Preconcentration and determination of As, Cd, Pb and Bi using different sample introduction systems, cloud point extraction and inductively coupled plasma optical emission spectrometry
Fernanda
dos Santos Depoi
,
Tiago
Charão de Oliveira
,
Diogo
Pompéu de Moraes
and
Dirce
Pozebon
*
Universidade Federal do Rio Grande do Sul, Instituto de Química, 91501-970, Porto Alegre, RS, Brazil. E-mail: dircepoz@iq.ufrgs.br; Fax: +55 33087304; Tel: +55 51 33087215
First published on 7th October 2011
Abstract
This study deals with the development of a method for As, Bi, Cd and Pb preconcentration and determination using cloud point extraction (CPE) and inductively coupled plasma optical emission spectrometry (ICP OES). Hydride generation, pneumatic nebulization and micronebulization/aerosol desolvation were investigated for introducing the surfactant rich phase into the ICP. O,O-Diethyldithiophosphate (DDTP) was used as complexant and octylphenoxypolyethoxyethanol (Triton X-114) as surfactant. The influence of concentration of HNO3, HCl, DDTP, Triton X-114, surfactant rich phase in methanol, reductant of As, and NaBH4 was evaluated. The enrichment factors obtained were 10, 18, 12 and 14 for As, Bi, Cd and Pb, respectively. The limits of detection (LODs) of As, Bi, Cd and Pb were 0.055, 0.063, 0.047 and 0.28 μg L−1, respectively. Precision and accuracy were assessed by analysis of certified enriched water (NIST 1643e), oyster tissue (NIST 1566b), tobacco leaves (CTA-OTL-1), bush branches and leaves (GBW 07602) and analyte spiking. Microwave-induced combustion (MIC), sonication, and acid digestion were used for sample preparation. The developed method was applied for extraction and determination of As, Bi, Cd and Pb in river water, wine, fertilizer and urine. Analyte recovery close to 100% and relative standard deviation (RSD) lower than 5% were observed.
1. Introduction
Matrix separation/analyte preconcentration has been used to reduce interference and also improve limits of detection (LODs).1,2 Among the different methods used for matrix separation/analyte preconcentration, cloud point extraction (CPE) is outstanding.2–7 Cloud point extraction is based on micelles formation and subsequent separation. Micellar aqueous solution is produced by addition of surfactant. The amount of surfactant added must be such to ensure the formation of micelle aggregates in the solution [the final surfactant concentration must exceed the critical micelle concentration (CMC)]. Once the surfactant concentration exceeds the CMC, the aqueous micellar solution separates into two isotropic phases: a surfactant-rich phase of small volume and a surfactant-poor phase of much higher volume (aqueous). The separation of the phases can be accelerated by increasing the temperature and addition of salt. Any component that binds to the micellar aggregate in solution can be extracted from the original solution and, therefore, be concentrated in the surfactant-rich phase.8 In the case of inorganic species, which are hydrophilic, complexing agents are used for producing hydrophobic species. These species produced are then extracted into micelles.2,9Reagents such as 8-hydroxyquinoline,3O,O diethyldithiophosphate (DDTP)1,7,10 and ammonium pyrrolidinedithiocarbamate (APDC)11 have been used as complexants. DDTP is quite stable in acid medium that is very important because samples are usually decomposed with acid.7,10 The octylphenoxypolyethoxyethanol (Triton X-114, a nonionic surfactant) has been widely used11,12 mainly because of the relatively low CPE temperature (between 22 and 25 °C) and low cost.
Analyte preconcentration using CPE can be performed with a small volume of surfactant, which is an advantage of the method. Complexant, pH, ionic strength, surfactant type and concentration, temperature, time of reaction and centrifugation have to be evaluated to make CPE successful. High electrolyte concentration and high acidity can prevent analyte preconcentration and/or CPE. Thus, previous sample preparation must be carefully evaluated.
Atomic absorption (AAS) techniques and inductively coupled plasma optical emission spectrometry (ICP OES) are usually employed for analyte detection after its preconcentration using CPE.1,13,14 However, CPE in conjunction with ICP OES and hydride generation (HG) has not been much investigated for hydride forming elements. Quite low LODs are expected, especially if an ICP OES spectrometer with axially viewed plasma is used for detection.
Due to viscosity and organic content of the surfactant-rich phase, which affects the plasma performance and stability, appropriate sample introduction systems and nebulizers are required. For example, flow injection (FI) was used for introducing a small volume of the surfactant-rich phase into plasma,3 or the analyte present in the surfactant-rich phase was retained in a column made with cotton, eluted with a mixture of nitric acid and propanol and subsequently introduced into plasma.13 Other examples are employment of free-clogging nebulizer14 or chemical vapour generation.5
Micronebulization/aerosol desolvation has not been investigated for introducing the surfactant rich phase into ICP. Aerosol desolvation promotes better sample transport efficiency to plasma and better sensitivity as a consequence. By using micronebulization/aerosol desolvation analysis of a very low amount of sample is possible.15,16 This is an advantage since the volume of the surfactant-rich phase is small. On the other hand, more critical effects of organics are expected because the amount of them introduced into the plasma increases due to the higher sample transport efficiency.
The potential of CPE for matrix separation/Cd, Pb, Bi and As preconcentration in different matrices followed by analyte detection using ICP OES is investigated in the present work. Micronebulization/aerosol desolvation and HG are proposed for introducing the surfactant-rich phase into plasma. Different procedures of sample preparation are used. In order to obtain a solution with low acid concentration, microwave induced combustion (MIC)17 is employed.
2. Material and methods
2.1. Instrumentation
An Optima 2000 DV-ICP OES spectrometer (PerkinElmer, Norwalk, CT, USA) was used. Argon (White Martins/Praxair) was used as plasma gas and auxiliary gas, whereas nitrogen with purity of 99.996% (White Martins/Praxair) was used as purging gas. The main instrumental parameters are summarized in Table 1.| Parameter | Arsenic | Cadmium and lead | Bismuth |
|---|---|---|---|
| Plasma power/W | 1500 | 1500 | 1400 |
| Plasma gas flow rate/L min−1 | 15 | 15 | 15 |
| Auxiliary gas flow rate/L min−1 | 0.2 | 0.2 | 0.2 |
| Nebulizer or carrier gas flow rate/L min−1 | 0.6 | 0.6 | 0.6 |
| Sample introduction | Hydride generation | GemCone; unbaffled cyclonic spray chamber | APEX-Q system; hydride generation |
| Spectral line/nm | 193.696 | 228.802 (Cd) and 220.353 (Pb) | 223.061 |
| Background correction | 2 points/peak | 2 points/peak | 2 points/peak |
| Signal processing (peak area) | 7 points/peak | 3 (Cd) and 7 (Pb) points/peak | 7 points/peak |
A home made HG system was hyphenated with ICP OES and used for As and Bi determination. This system is described elsewhere.5 It consists basically of a confluence and a gas liquid separator. In this system, solutions were transported and mixed using the peristaltic pump of the ICP OES spectrometer. The flow rate of sample, HCl and NaBH4 solutions were 1.3, 1.8 and 1.3 mL min−1, respectively. The pneumatic nebulizer used (GemCone)18 is considered free-clogging and suitable for viscous solutions or having high content of dissolved solids. An APEX-Q system (ESI, USA) with aerosol desolvation was used. Solutions were aspirated through a PFA microconcentric nebulizer fitted into a cyclonic spray chamber that was heated at 140 °C and then transported to a Peltier-cooled multipass condenser where the temperature was set as 2 °C. Partial solvent removal occurs in this system and sample transport efficiency is about 30%. The efficiency of this system is presented elsewhere.15,16
A heating block (TE-007D Tecnal, Brazil) was used for the fertilizer sample decomposition. A microwave oven Multiwave 3000 (Anton Paar) equipped with quartz vessels was used for MIC. A water bath with temperature control was used as a source of heating and assists CPE while a centrifuge was used for separation of the aqueous and surfactant-rich phases. A Hydraulic Press (15 ton) was used for pellets preparation used in MIC.
2.2. Reagents, solutions and materials
All chemicals were of analytical-grade. Water purified (to 18.2 MΩ cm) in a Milli-Q system (Millipore) was used to prepare all reagents, solutions and samples. Nitric acid (65% m/m), HCl (37% m/m), H2O2 (30% m/m) and CH3OH (all from Merck) were used. The HNO3, HCl and CH3OH used were further purified by sub-boiling distillation (a Milestone duo PUR 2.01E system was used). Sodium tetrahydroborate (NaBH4, Vetec, Brazil) was employed for As and Bi determination using HG. DDTP [(C2H5O)2P(S)SNH4] from Aldrich and Triton X-114 from Sigma were used for As, Cd, Pb and Bi preconcentration. A 5.0% (m/v) DDTP stock solution was prepared by the dissolution of the reagent in water. A 5.0% (m/v) Triton X-114 stock solution was prepared by weighing 2.5 g of the reagent in a polypropylene vial and adding 50 mL of water. Antifoam Y-30 from Sigma-Aldrich was used to reduce foam production in determinations using HG. Potassium iodine (KI) and ascorbic acid (C6H8O6) from Vetec were used for As reduction. Solutions of As, Cd, Pb and Bi were prepared in 0.14 mol L−1 HNO3 by serial dilution of mono-element stock solutions (Titrisol, Merck) containing 1000 mg L−1 of the analyte. The calibration solutions concentration and respective acid concentration varied according to the analyte as will be seen later. The calibration solutions were also submitted to CPE in the same way as that of samples.A 6.0 mol L−1 ammonium nitrate (Merck) solution was used as igniter for MIC. A small disc (15 mm of diameter and mass of 12 mg) of paper with low ash content was also used to aid the combustion process. A more detailed procedure of sample preparation using MIC is described elsewhere.14
2.3. Samples and sample preparation
Samples of the following certified reference materials (CRMs) were analyzed: water (NIST 1643e) and oyster tissue (NIST 1566b) from the National Institute of Standards and Technology, bush branches and leaves (GBW 07602) from the Institute of Nuclear Research of China, and oriental tobacco leaves (CTA-OTL-1) from the Institute of Nuclear Chemistry and Technology of Poland. The solid samples of the CRMs were decomposed using MIC. Approximately 300 mg of powder sample were pressed into pellets, weighed directly on filter paper and then placed on a quartz holder positioned inside a quartz vessel to which 6 mL of absorbing solution (0.4 mol L−1 HNO3) was previously added. Then, 50 μL of ammonium nitrate solution was immediately added to the paper. After closing the quartz vessels and placing them in the rotor they were pressurized with oxygen at 20 bar for 2 min and the rotor placed inside the microwave oven. Next, the mixtures inside the vessels were irradiated by microwave for 60 s at 1400 W followed by cooling for 20 min. The resultant solutions were transferred to graduated polypropylene vials and the volume completed to 25 mL using water. A sample of bush branches and leaves (GBW 07602) was also prepared by sonication; aliquots of 300 mg in 25 mL of 2.4 mol L−1 HCl were sonicated for 30 s at 80 W using a probe. An ultrasonic processor (Unique, Brazil) equipped with a 4 mm diameter titanium tip was used. The sonicated mixture was subsequently centrifuged for 10 min at 3200 rpm. Aliquots of the supernatant were then submitted to CPE.Fertilizer and white wine purchased in the local market, river water (from Rio Guaíba, RS, Brasil) and urine (from a volunteer) were analyzed. Aliquots of 0.100 g of the fertilizer (previously pulverized in agate mortar) were weighed and transferred to PTFE flasks to which 1.0 mL HNO3 and 1.0 mL H2O2 were added. The mixture was left to stand for a period of 12 h. Subsequently, the flasks were closed with screw caps and the mixture heated at 100 °C for 4 h. After cooling at room temperature, the obtained solutions were transferred to graduated polypropylene vials and the volume completed to 25 mL using water.19 The river water was collected in a cleaned polyethylene bottle and just filtered (a Whatman filter paper for fast filtration was used). The urine and wine samples were sonicated with a probe for 30 s at 80 W. Then, they were ten-fold diluted with water before being submitted to CPE. Analyte recovery tests were performed in order to verify the accuracy and precision of the method for wine, river water and fertilizer analysis. The liquid samples were spiked before being submitted to CPE whereas the fertilizer was spiked before decomposition.
2.4. Cloud point extraction
Aliquots of sample solution ranging from 1 to 7 mL were transferred to graduated polypropylene vials. For As determination, KI and C6H8O6 (ascorbic acid) were added to the solution in order to obtain only As(III) species, which reacts with DDTP. After addition of KI and C6H8O6 the mixture was allowed to stand for 30 minutes. Next, DDTP and Triton X-114 solutions were added and the volume of the mixture was completed to 14 mL using water and/or acid solution. The optimal concentrations of all reagents in the final mixture were investigated and they are summarized in Table 2. The mixture was heated in a water bath in order to accelerate the separation of the phases, centrifuged at 3200 rpm for 10 min and then cooled in an ice bath for 10 min. The aqueous phase was removed by inversion of the vial and the surfactant-rich phase was subsequently taken by using a Pasteur pipette. The final volume of the surfactant-rich phase ranged from 50 to 150 μL. Different amounts of methanol were added to the surfactant-rich phase, depending on the sample introduction system used. Dilution with methanol was followed by addition of 1.0 mL of 0.60 mol L−1 HCl for As, 0.5 mL of 0.70 mol L−1 HNO3 or 1.0 mL of 0.5 mol L−1 HCl for Bi, and 1.0 mL of 0.70 mol L−1 HNO3 for Cd and Pb.| Condition | As | Cd and Pb | Bi |
|---|---|---|---|
| DDTP concentration (% m/v) | 0.25 | 0.20 | 0.15 |
| Triton X-114 (% m/v) | 0.05 | 0.15 | 0.15 |
| Extraction medium and concentration/mol L−1 | HCl/0.96 | HNO3/0.28 | HNO3/0.40 |
| Temperature/°C | 50 | 50 | 50 |
| Period of heating/min | 20 | 20 | 20 |
| Surfactant rich-phase volume/μL | 50 | 150 | 150 |
| Volume of methanol added to the surfactant rich-phase/μL | 100 | 50 | 100 |
| HCl concentration for hydride generation/mol L−1 | 4.8 | — | 3.0 |
| NaBH4 concentration (% m/v) | 0.5 | — | 0.5 |
3. Results and discussion
3.1. Analyte preconcentration and detection
![Influence of the reagents concentration on As [20 μg L−1 of As(v)] pre-concentration in (a) and (b). KI: potassium iodine; aa: ascorbic acid. Conditions: 0.20% m/v DDTP, 0.05% m/v Triton X-114 and 100 μL of methanol in (a) and in (b) 0.05% m/v Triton X-114, 0.96 mol L−1 HCl, 0.5% m/v KI/aa and 100 μL of methanol. Hydride generation conditions: 0.5% m/v NaBH4 and 4.0 mol L−1 HCl.](/image/article/2012/AY/c1ay05246b/c1ay05246b-f1.gif) | ||
| Fig. 1 Influence of the reagents concentration on As [20 μg L−1 of As(V)] pre-concentration in (a) and (b). KI: potassium iodine; aa: ascorbic acid. Conditions: 0.20% m/v DDTP, 0.05% m/v Triton X-114 and 100 μL of methanol in (a) and in (b) 0.05% m/v Triton X-114, 0.96 mol L−1 HCl, 0.5% m/v KI/aa and 100 μL of methanol. Hydride generation conditions: 0.5% m/v NaBH4 and 4.0 mol L−1 HCl. | ||
With respect to As hydride generation, better sensitivity was observed for 4.8 mol L−1 HCl and 0.5% (m/v) NaBH4. Aqueous solution not submitted to CPE was also analyzed just for comparison. The best conditions for As hydride generation in solution not submitted to CPE mismatched with those for solution submitted to CPE. The differences may be due to the different medium and also foaming production into the gas–liquid separator. Anyway, this inconvenience did not preclude obtaining accurate results because the calibration solutions were also subjected to CPE.
The surfactant-rich phase was also introduced into plasma by the micronebulization/aerosol desolvation system employed in the present work. However, the analyte signal did not stabilize and the LOD was worse than that obtained by using HG. Therefore, HG was selected for As determination.
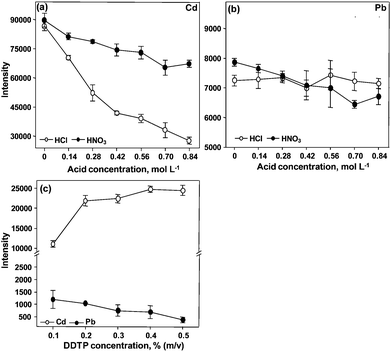 | ||
| Fig. 2 Influence of the reagents concentration on pre-concentration of Cd and Pb using CPE. A solution containing 20 μg L−1 of Cd and Pb was used. Conditions: 0.30% m/v DDTP, 0.15% m/v Triton X-114 and 50 μL of methanol in (a) and (b); 0.15% m/v Triton X-114, 0.28 mol L−1 HNO3 and 50 μL of methanol in (c). | ||
With respect to Triton X-114, the highest signals are observed when the surfactant concentration is 0.15% (m/v). The signals of Cd and Pb decrease in the presence of Triton X-114 higher than 0.15% (m/v). This behavior had already been observed by other authors, for Cd and Pb preconcentration in blood12 and seawater7 prior to the analytes determination using ETAAS and ICP-MS, respectively. The influence of the DDTP concentration is shown in Fig. 2(c). It can be seen that the signal of Cd increases with the increase of DDTP concentration up to 0.4% (m/v). On the other hand, the signal of Pb decreases with the increase of the complexant concentration. Thus, keeping in mind the possibility of measuring Pb and Cd simultaneously, 0.20% (m/v) DDTP was chosen for both analytes as a compromise condition. In order to reduce the viscosity of the surfactant-rich phase, 50 μL of methanol were added to it. A minimum volume of methanol was added considering the solvent effects in the plasma.
Micronebulization/aerosol desolvation was also investigated for Cd and Pb determination. The sensitivity was improved, but precision was not acceptable (RSD around 30%). This system was then not employed for further Cd and Pb determination in the surfactant-rich phase due to the bad precision observed. The main reason was the effect of the surfactant and methanol that were present in the plasma, since very good precision (RSD lower than 3%) was observed for Cd and Pb in aqueous solution by using the same nebulizer. In this case the LODs of Cd and Pb were 0.07 and 0.89 μg L−1, respectively. The effect of surfactant and methanol is not the same for all elements. The same effect observed for Cd and Pb was not observed for Bi, as will be seen later.
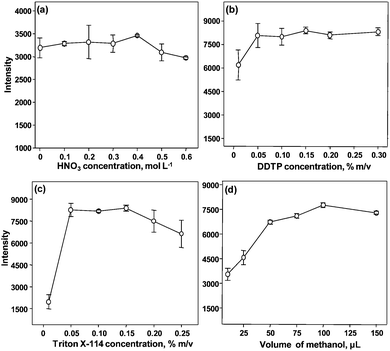 | ||
| Fig. 3 Influence of the reagents concentration on preconcentration of Bi using CPE. A solution containing 20 μg L−1 of Bi was used. Conditions: 0.30% m/v DDTP, 0.15% m/v Triton X-114 and 50 μL of methanol in (a); 0.15% m/v Triton X-114, 0.40 mol L−1 HNO3 and 50 μL of methanol in (b); 0.15% m/v DDTP, 0.40 mol L−1 HNO3 and 50 μL of methanol in (c); 0.15% m/v DDTP, 0.15% m/v Triton X-114 and 0.40 mol L−1 HNO3 in (d). | ||
It was observed that the concentration of the surfactant used has a great influence on Bi preconcentration. According to Fig. 3(c), the appropriate concentration ranges from 0.05 to 0.15% m/v Triton X-114. This is in accordance with results published by other authors23 who used Triton X-114 and dithizone as complexant of Bi. With respect to the amount of methanol added to the surfactant-rich phase, it was less critical in comparison to the conventional pneumatic nebulization. As shown in Fig. 3(d), the highest signal of Bi was obtained for 100 μL of methanol.
Hydride generation was investigated for Bi determination in the surfactant-rich phase. For this, the influence of HCl and NaBH4 was evaluated. As shown in Fig. 4, sensitivity is better for HCl (3.0 mol L−1). Sensitivity increased with the increase in the NaBH4 concentration but the NaBH4 concentration was fixed at 0.5% (m/v) due to excessive foam production into the gas–liquid separator. Similar LODs were obtained using micronebulization/aerosol desolvation or HG (see Table 3). Therefore, micronebulization/aerosol desolvation or HG can be used for Bi determination in the surfactant-rich phase. The main advantage of micronebulization/aerosol desolvation was lower consumption of reagents in comparison to HG.
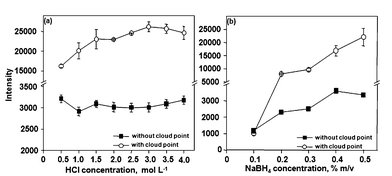 | ||
| Fig. 4 Influence of the reagents concentration on hydride generation of Bi using CPE. A solution containing 15 μg L−1 of Bi was used. Conditions: 0.40 mol L−1 HNO3, 0.15% m/v DDTP, 0.15% m/v Triton X-114 and 100 μL of methanol. | ||
| Element/system | Calibration curve/μg L−1 | Equation | EF | LOD/μg L−1 | LODa/μg g−1 |
|---|---|---|---|---|---|
| a 300 mg of sample in 30 mL of solution and four fold dilution were taken into account; EF: enrichment factor; LOD: limit of detection. | |||||
| As/CPE-HG | 0.50–10.0 | y = 2157x + 2.7 | 10 | 0.055 | 0.022 |
| As Hydr. generation | 2.0–20.0 | y = 205.3x − 6.0 | — | 0.15 | 0.062 |
| Bi/CPE Micr./desolvation | 0.5–10.0 | y = 747x + 144 | 18 | 0.063 | 0.026 |
| Bi Neb/desolvation | 5.0–25.0 | y = 41.2x + 23 | — | 0.46 | 0.19 |
| Bi/CPE-HG | 0.5–10.0 | y = 4442x + 305 | 7 | 0.057 | 0.024 |
| Bi Hydr. generation | 5.0–25.0 | y = 680x + 25 | — | 0.13 | 0.050 |
| Cd/CPE nebulization | 1.0–15.0 | y = 7128x + 1382 | 12 | 0.047 | 0.018 |
| Cd nebulization | 5.0–50.0 | y = 616x + 13 | — | 0.18 | 0.072 |
| Pb/CPE nebulization | 5.0–25.0 | y = 487x + 45 | 14 | 0.28 | 0.12 |
| Pb nebulization | 15.0–50.0 | y = 35.6x − 12 | — | 4.0 | 2.4 |
3.2. Figures of merit
The figures of merit of the proposed method and calibration curve parameters are summarized in Table 3. The LODs were calculated according to IUPAC (International Union of Pure and Applied Chemistry) recommendations. The LOD was obtained from b + 3s; b is the mean concentration of 10 consecutive measurements of the blank and s is the standard deviation. The blank for each element underwent the same procedure of the samples and calibration solutions. The enrichment factor (EF) was calculated by the ratio of the slope of calibration curves obtained (with and without analyte pre-concentration) for each system used for introducing the surfactant-rich phase into plasma. The surfactant-rich phase (volume ranging from 50 to 150 μL) needed to be diluted (see Section 2.4) due to the high viscosity or foam production into the gas liquid separator, which decreased the EF. Despite the dilution involved, the LODs found in the present work are of the same order of magnitude or lower than values reported in the literature regarding to As, Cd, Pb and Bi preconcentration using CPE, FAAS or ICP OES.3,63.3. Samples analysis
The accuracy and precision of the developed method were evaluated by the analysis of certified samples and recovery tests. As can be seen in Table 4, the results obtained for Cd and Pb are in agreement with the certified values (at 95% confidence level) for all analysed samples. This demonstrates that microwave induced combustion (MIC) is adequate for preparing samples of vegetal and animal tissues for Cd and Pb preconcentration and determination using CPE and ICP OES. The acid concentration in the resultant sample solution is low and, therefore, favourable to Cd and Pb preconcentration. Bismuth was detected only in certified water and the concentration found was in agreement with the certified value. This element was not detected in bush branches and leaves whose Bi concentration informed on the certificate is lower than the LOD of Bi (0.026 μg g−1).| Sample | Analyte | Certified/μg g−1 | Found/μg g−1 |
|---|---|---|---|
| a In μg L−1; sample preparation. b MIC. c Sonication; nd: not detected. | |||
| NIST 1566b (oyster tissue) | As | 7.65 ± 0.65 | 7.30 ± 0.28 |
| Bi | — | — | |
| Cd | 2.48 ± 0.08 | 2.42 ± 0.11 | |
| Pb | 0.308 ± 0.009 | 0.320 ± 0.010 | |
| GBW 07602 (bush branches and leaves) | As | 0.950 ± 0.080 | 0.270 ± 0.028,b 1.022 ± 0.045c |
| Bi | 0.022 | nd | |
| Cd | 0.14 ± 0.01 | 0.13 ± 0.01 | |
| Pb | 6.5 ± 0.9 | 6.3 ± 0.1 | |
| CTA-OTL-1 (oriental tobacco) leaves | As | 0.539 ± 0.060 | 0.458 ± 0.030 |
| Bi | — | — | |
| Cd | 1.12 ± 0.12 | 1.01 ± 0.01 | |
| Pb | 4.91 ± 0.80 | 4.04 ± 0.08 | |
| NIST 1643e enriched watera | As | 60.45 ± 0.72 | 62.72 ± 1.98 |
| Bi | 14.09 ± 0.15 | 14.62 ± 0.65 | |
| Cd | 6.568 ± 0.073 | 6.305 ± 0.007 | |
| Pb | 19.63 ± 0.21 | 19.32 ± 0.54 |
With respect to As, it is observed that the mean concentration found in tobacco leaves (CTA-OTL-1) is lower than the certified value. Nevertheless, the concentration range (mean and standard deviation) is not different for a 95% confidence level. The concentration of As found for bush branches and leaves (GBW07602) submitted to microwave induced combustion (MIC) is different from the certified value. But the concentration found agrees with the certified value if the sample is sonicated and submitted to CPE. In this step of the work it was concluded that additional studies are necessary for the decomposition of vegetal samples using MIC with the aim of As determination.
The results obtained for urine, white wine, river water and chemical fertilizer samples are presented in Table 5. Arsenic was detected in white wine and fertilizer, while Cd and Pb were detected only in fertilizer. Bismuth was not detected in any sample analyzed. Despite the fact that wine, urine and fertilizer have complex matrices, good recoveries were found for all analytes, indicating efficient matrix separation. This also demonstrates that the proposed method can be used for the determination of As, Bi, Cd and Pb in different matrices.
| Sample | Analyte | Found/μg L−1 | Spiked/μg L−1 | Found/μg L−1 | Recovery (%) |
|---|---|---|---|---|---|
| a In μg g−1 (nd: not detected). | |||||
| Urine | As | nd | 5.00 | 5.48 ± 0.30 | 110 |
| Bi | nd | 10.0 | 9.74 ± 0.03 | 97 | |
| Cd | nd | 5.00 | 4.71 ± 0.08 | 97 | |
| Pb | nd | 10.0 | 10.1 ± 0.18 | 100 | |
| White wine | As | 9.51 ± 0.11 | 5.00 | 15.0 ± 0.28 | 110 |
| Bi | nd | 10.0 | 9.31 ± 0.15 | 93 | |
| Cd | nd | 5.00 | 4.72 ± 0.08 | 94 | |
| Pb | nd | 10.0 | 9.61 ± 0.15 | 96 | |
| River water | As | nd | 5.00 | 5.15 ± 0.07 | 103 |
| Bi | nd | 10.0 | 9.61 ± 0.26 | 96 | |
| Cd | nd | 5.00 | 4.91 ± 0.03 | 98 | |
| Pb | nd | 10.0 | 10.1 ± 0.18 | 101 | |
| Fertilizera | As | 0.67 ± 0.04 | 1.25 | 1.80 ± 0.05 | 91 |
| Bi | nd | 2.5 | 2.31 ± 0.30 | 92 | |
| Cd | 3.95 ± 0.54 | 2.5 | 6.58 ± 1.75 | 105 | |
| Pb | 0.71 ± 0.26 | 2.5 | 3.21 ± 0.35 | 100 |
4. Conclusions
The results obtained demonstrated that matrix separation/analyte preconcentration using CPE allowed the determination of low concentrations of As, Bi, Cd and Pb in different matrices. Arsenic and Bi at ng L−1 level can be determined using CPE for element preconcentration and HG or micronebulization/aerosol desolvation for introducing the surfactant-rich phase in the plasma. Sensitivities for Bi, Cd and Pb improved through the use of micronebulization/aerosol desolvation. However, the precision for Cd and Pb was worse and no improvement of the LODs was observed for these two elements in the surfactant rich phase in comparison to conventional pneumatic nebulization. As known,9,22 the complexation of the investigated elements with DDTP occurs in acidic medium (HCl or HNO3). However, in the present work it was observed that for offline preconcentration of Cd and Pb the acid concentration must be low. In this sense, the use of MIC has proved to be advantageous, since acid concentration in the final solution is low in comparison to conventional decomposition in a microwave oven.Acknowledgements
Fernanda dos Santos Depoi would like to thank CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior from Brazil) for the scholarship received.References
- L. M. Coelho and M. A. Z. Arruda, Spectrochim. Acta, Part B, 2005, 60, 743 CrossRef.
- E. K. Paleologos, D. L. Giokas and M. I. Karayannis, TrAC, Trends Anal. Chem., 2005, 24, 426 CrossRef CAS.
- M. Sun and Q. Wu, J. Hazard. Mater., 2011, 192, 925 CrossRef.
- D. Pozebon, V. L. Dressler, J. A. Gomes Neto and A. J. Curtius, Talanta, 1998, 45, 1167 CrossRef CAS.
- F. S. Depoi, F. R. S. Bentlin and D. Pozebon, Anal. Methods, 2010, 2, 180 RSC.
- J. Manzoori and A. B. Tabrizi, Anal. Chim. Acta, 2002, 470, 215 CrossRef CAS.
- M. A. M. Silva, V. L. A. Frescura and A. J. Curtius, Spectrochim. Acta, Part B, 2000, 55, 803 CrossRef.
- C. D. Stalikas, TrAC, Trends Anal. Chem., 2002, 21, 343 CrossRef CAS.
- W. L. Hinze and E. Pramauro, Crit. Rev. Anal. Chem., 1993, 24, 133 CrossRef CAS.
- E. L. Seibert, V. L. Dressler, D. Pozebon and A. J. Curtius, Spectrochim. Acta, Part B, 2001, 56, 1963 CrossRef.
- A. Tang, G. S. Ding and X. Yan, Talanta, 2005, 67, 942 CrossRef CAS.
- D. L. G. Borges, M. A. M. S. Veiga, V. L. A. Frescura, B. Welz and A. J. Curtius, J. Anal. At. Spectrom., 2003, 18, 501 RSC.
- Y. Yamini, M. Faraji, S. Shariari, R. Hassani and M. Ghambarian, Anal. Chim. Acta, 2008, 612, 144 CAS.
- S. Shariati, Y. Yamini, M. Khalili and K. Zanjani, J. Hazard. Mater., 2008, 156, 583 CrossRef CAS.
- V. L. Dressler, D. Pozebon, A. Matusch and J. S. Becker, Int. J. Mass Spectrom., 2007, 266, 25 CrossRef CAS.
- D. Pozebon, V. L. Dressler, J. S. Becker, A. Matusch, M. Zoriyd and J. S. Becker, J. Anal. At. Spectrom., 2008, 23, 1281 RSC.
- M. F. Mesko, D. P. Moraes, J. S. Barin, V. L. Dressler, G. Knapp and E. M. M. Flores, Microchem. J., 2006, 82, 183 CrossRef CAS.
- J. Nölte, ICP Emission Spectrometry: a Practical Guide, Willey–VCH Verlang GmbH & Co. KGaA, Weinheim, Germany, 2003 Search PubMed.
- S. M. Macedo, R. M. de Jesus, K. S. Garcia, V. Hatje, A. F. S. Queiroz and S. L. C. Ferreira, Talanta, 2009, 80, 974 CrossRef CAS.
- J. Frank, M. Krachler and W. Shotyk, Anal. Chim. Acta, 2005, 530, 307 CrossRef CAS.
- D. Q. Hung, O. Nekrassova and R. G. Compton, Talanta, 2004, 64, 269 CrossRef CAS.
- G. Cote and D. Bauer, Anal. Chem., 1984, 56, 2153 CrossRef CAS.
- F. Shemirani, M. Baghdadi, M. Ramezani and M. R. Jamali, Anal. Chim. Acta, 2005, 534, 163 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |