A simple system for the measurement of the distribution of activities of individual molecules of E. coli β-galactosidase
Douglas B.
Craig
*
Chemistry Department, University of Winnipeg, Winnipeg, Manitoba, Canada R3B 2E9. E-mail: d.craig@uwinnipeg.ca; Fax: (+204) 774 2401; Tel: (+204) 786 9731
First published on 15th December 2011
Abstract
A simple system capable of measuring the distribution of single molecule activities of a population of E. coli β-galactosidase was developed. This system utilized a modified disposable syringe, a narrow bore capillary tube and an in-laboratory constructed laser-induced fluorescence detection system. Once started, the experiment can continue to collect data without any operator input for more than 24 h. The relative catalytic rates of 138 individual molecules of the enzyme were determined. The distribution of activities was found to be indistinguishable from that reported using more complicated methodologies.
Classical enzymology studies involve measurements made on samples containing very large numbers of individual enzyme molecules. Data obtained represents average values for the ensemble, and often does not take into account that a fraction of the population may be denatured or otherwise inactive. The development of techniques which are capable of measuring the properties of individual enzyme molecules have permitted the observation of properties otherwise obscured by large scale averaging. These include counting of the number of active molecules, measurement of the differences in properties of the previously assumed to be identical molecules, and for those techniques that provide time resolution, measurement of the variation in activity of individual enzyme molecule over time.
The first report of the assaying of an individual enzyme molecule was that of Rotman.1 The assay mixture was dispersed into fine droplets and coated in oil to prevent evaporation. The droplets were incubated for 15 h and the product formed detected using fluorescence microscopy. From the droplet volume and enzyme concentration, and since the number of enzyme molecules per droplet was determined by random sampling statistics, the system was set up such that the large majority of droplets were predicted to contain either zero or one enzyme molecule. Signal obtained from a particular droplet that exceeded the background was attributed to the activity of a single enzyme molecule.
The study of individual enzyme molecules reemerged when Yeung's group introduced a capillary electrophoresis laser-induced fluorescence based method used in the assaying of lactate dehydrogenase,2 followed by Dovichi's group using a similar methodology with alkaline phosphatase.3 In this method a 40–70 cm long narrow bore capillary was filled with dilute enzyme and substrate and incubated. Enzyme concentration was such that on average 5–20 molecules were present within the capillary, and were on average several centimeters apart. Due to the relative short incubation time, there was insufficient time for the product formed from one enzyme molecule to diffuse to and mix with that of another. Instead localized pools of product, each due to the actions of an individual enzyme molecule, were formed. Post-incubation electrophoresis was used to mobilize these pools past the detector.
In addition to the capillary electrophoresis approach, Yeung's group developed a fluorescence microscopy protocol for the assaying of individual molecules of lactate dehydrogenase.4Pores were created in membranes or silica using photolithography, forming femtoliter vessels. These vessels were filled with the assay mixture. Similarly to Rotman, enzyme concentration was such that usually either zero or one LDH molecule was present per vessel. As the sample incubated it was excited using a laser and signal collected with a CCD camera. This permitted continuous monitoring of the accumulation of the fluorescent product NADH. The fluorescence microscopy approach has been adopted by other groups, using both free and tethered enzyme molecules and ‘real time’ measurements.5,6
One shortcoming of the capillary electrophoresis approach is that in a given experiment only 5–20 enzyme molecules are assayed. Walt's group used a different approach which allowed the monitoring of hundreds of individual enzyme molecules per experiment.7,8 In this approach bundles of small diameter optical fibers were cut, polished and etched with HCl, producing wells of 40 fL volume. Assay mixture was placed onto a cleaned silicon gasket which was pushed up against the end of the fiber bundle, filling up and sealing the reaction chambers. Alternately, the wells had biotin covalently attached and this was used to capture streptavidin labeled enzyme.9 The samples were excited through the optical fibers using a short arc mercury lamp. Fluorescence was collect though total internal reflectance along the same optical fibers and images were taken using a CCD camera every 15 s.
The major finding of these studies was that individual molecules of a given enzyme present heterogeneous properties. Catalytic rate, in terms of both Km10 and Vmax,3 have been shown to differ widely between individual molecules. Additionally, activation energy of catalysis,3 and electrophoretic mobility11 have been shown to be heterogeneous. Furthermore, catalytic rate has been found to vary over time for an individual molecule.4–6,12,13 The structural basis for these differences is not clear, although conformational differences,2 differential post-translational modification3 and random errors in transcription and translation11 have been suggested.
One drawback of previous methods is that they involve experimental setups that can be relatively complex to design and construct, as well as being time consuming to obtain proficiency with. Here a simplified system is described which allows for the measurement of the distribution of catalytic activities of individual molecules of the enzyme β-galactosidase. Once an experiment was initiated, the system allowed for the continuous collection of data from a sample without any input from the operator for a period of longer than 24 h.
Materials and methods
Chemicals
7-hydroxy-9H-(1,3-dichloro-9,9-dimethylacridin-2-one) (DDAO) and 9H-(1,3-dichloro-9,9-dimethylacridin-2-one-7-yl) β-D-galactopyranoside (DDAO-gal) were obtained from Invitrogen/Molecular Probes (Eugene, OR). Hydrolysed casein was from Difco (BD, Sparks, MD). Isopropyl-β-D-thiogalactoside (IPTG) and all other reagents were from Sigma (St. Louis, MO).β-galactosidase
Wild-type E. coli (American Type Culture Collection strain 35321) was grown for 20 h at 37 °C in 12.29 g/L M9 minimal salts containing 1% (w/v) glycerol, 0.2% (w/v) hydrolyzed casein, 1 mM MgCl2, 50 μM Fe2SO4, and 50 μM ZnCl2. β-Galactosidase expression was induced by the addition of IPTG to a final concentration of 250 μM. After a further 1 h incubation period at 37 °C, the culture was centrifuged at 10,000 × g for 10 min at 5 °C. The pellet was re-suspended in approximately 500 μL of 100 mM HEPES (pH 7.3) containing 2 mM MgCl2 and 2% (v/v) protease inhibitor cocktail (no metal chelators) and frozen in N2(l). To lyse the cells, the sample was ground, briefly thawed and then refrozen. After five freeze/grind/thaw cycles the cold slurry was diluted with 3 ml of cold 100 mM HEPES (pH 7.3) containing 2mM MgCl2 and 2% (v/v) protease inhibitor cocktail (no metal chelators), and then centrifuged for 1 min at 10,000 × g to pellet cellular debris. The supernatant was passed through a sterile 0.45 μm filter and the filtrate was diluted with an equal volume of sterile glycerol and stored at −20 °C.Syringes
1 mL disposable plastic syringes (BD, Franklin Lakes, NJ) were modified for use in pressure injection of sample into capillaries. This modification was not novel and has been used in my laboratory, and others, for more than 15 years, however it has not been published previously. A disposable glass 10μL pipet (VWR, Mississauga, ON) was placed, approximately 2 cm from one end, into a flame and pulled apart using tongs, resulting in the tapering of the pipet. The capillary was threaded into the 2–3 cm long piece of pipet from the non-tapered end until it reached the point where it became wedged into the inside of the tapered end. The tapered end was then cut approximately 1 mm past the end of the capillary. The capillary was removed and the piece of pipet was placed into the end of syringe, with the tapered end inside and the non-tapered end hanging outside. The end of the plastic syringe was then placed in a flame and melted and then cooled such as to secure the piece of pipet in place (Fig. 1). A modified syringe can be routinely prepared in approximately 1–2 min. | ||
| Fig. 1 Photograph of a modified syringe being used to inject the sample into the capillary. | ||
Sample
1.48 μL of 27 mM DDAO-gal was added to 198.5 μL of 50 mM HEPES (pH 7.3) containing 1 mM MgCl2. In order to remove DDAO which is present as an impurity, the solution was washed by addition of 200 μL of toluene followed by mixing on a vortex for 1 min. The phases where separated by centrifugation for 30 s at approximately 5000 × g. The aqueous phase was then washed a second time with toluene. 125 μL of this substrate solution was then added to 370 μL of 50 mM HEPES (pH 7.3) containing 1 mM MgCl2. To this was added 5 μL of 105-fold diluted stock enzyme in 50 mM HEPES (pH 7.3) containing 1 mM MgCl2. Sample preparation requires approximately 3–4 min.Instrument
One end of a 100 cm long, 145 μm outer diameter, 5 μm internal diameter fused-silica capillary (Polymicro Technologies, Phoenix, AZ) was wedged into a modified syringe and the other end, from which approximately 1 mm of the polyimide coating was removed by flame, was placed in the detector. 200 μL of sample was added to the end of the syringe and the plunger pushed down such that the volume of air above the sample was reduced to approximately 20% of its original amount. The plunger of the syringe was pushed down by hand and held in place with the use of a 3-prong clamp (Fig. 1) and placed in an icebath such that the entire syringe and approximately 10 cm of capillary length was submerged. The detection end of the capillary was placed in the sheath flow cuvette (250 × 250 μm inner bore, Hellma, Concord ON) of a low-cost in-laboratory constructed laser-induced fluorescence detector that has been described previously.14 The 10 mW output of a 633-nm HeNe laser (Melles Griot, Nepean, ON) was focused with a 6.3X, 0.2 N.A. microscope objective (Melles Griot) approximately 10 μm below the end of the capillary. Emission was collected at 90° using a 60X 0.7 N.A. microscope objective (Universe Kogaku, Oyster Bay, NY), passed through a 660Df10 filter (Omega Optical, Brattleboro, VT) and onto a photomultiplier tube (PMT, Hamamatsu model 1477, Bridgewater, NJ). The analog signal was digitized using a Pentium 4 computer through a PCI-MIO-16XE I/O board utilizing LabView™ software (National Instruments, Austin, TX) at 10 Hz. The same board was used to control the PMT bias. Data was analyzed and peaks integrated using Igorpro™ software (Wavemetrics, Lake Oswego, OR).Results and discussion
In the assay, substrate containing very dilute enzyme was continuously pressure injected into a narrow bore capillary. Enzyme concentration was such that on average approximately 3–4 molecules of enzyme were travelling down the capillary at once. Since the capillary was 100 cm long, on average the molecules were decimeters apart. As an individual enzyme molecule traversed the capillary it formed product which travelled along with it. The relatively large distance between enzymes prevented product formed by one enzyme from diffusing to and mixing with that of another during the 40–45 min it took to travel the length of the capillary. Instead localized pools of product were obtained. As these pools migrated past the detector peaks were observed. Each peak represents the product formed by a single enzyme molecule. A portion of the signal output obtained is show in Fig. 2. Fig. 3 depicts the distribution of relative peak areas of 138 molecules assayed in a single run. This distribution is indistinguishable from that reported previous for E. coli β-galactosidase using both the capillary electrophoresis approach15 and that used by Walt.8 In this study wild-type E. coli was cultured in the laboratory and a crude homogenate prepared. Alternatively the enzyme can be obtained commercially from several different sources, including Sigma, at minimal cost.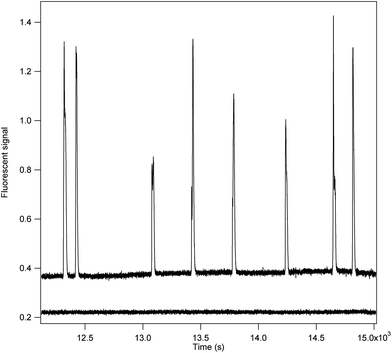 | ||
| Fig. 2 The signal output from the incubation of 8 molecules of E. coli β-galactosidase is shown. | ||
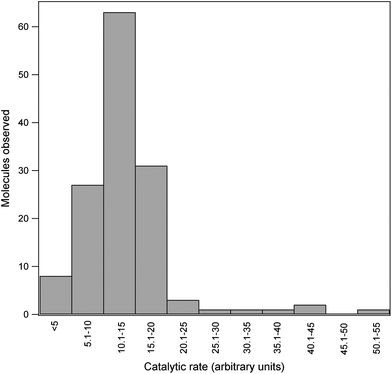 | ||
| Fig. 3 The distribution of the relative activities of 138 molecules of E. coli β-galactosidase is shown. | ||
The substrate DDAO-gal contains the product DDAO as an impurity and this was removed by washing with toluene immediately prior to use. However, DDAO-gal is slowly and non-enzymatically hydrolyzed to form DDAO, which resulted in a continuous rise in the baseline over time. Moreover, the enzyme present in the sample also continuously produces DDAO within the syringe, further increasing the background signal. This resulted in deterioration of the signal-to-noise ratio over time. In order to reduce the rate of the non-enzymatic formation of DDAO, the syringe was placed within an ice bath. This resulted in a greatly reduced rate of increase in the background signal and permitted extension of the useable assay time from 3–5 h to the 29 h used. Initially the signal-to-noise ratio for most peaks was >100 and remained >50 by the end of the 29 h incubation period used.
The number of peaks observed per unit time was found to decrease substantially over the 29 h period of the assay. One possible explanation is denaturation of the enzyme, however this was not observed in the capillary electrophoresis continuous flow assay which used the same assay mixture, albeit run times used were much shorter. Since in this assay the syringe was incubated in an ice bath, one possible explanation is a decrease in enzyme concentration over time due to absorption to surfaces in the syringe. Regardless, there was no obvious selective loss of more or less active enzyme molecules.
The time required to traverse the capillary, which can be measured by the time after the start of the experiment at which there is a sharp increase in the background signal due to the substrate starting to elute, was 40–45 min. Since the pressure applied by the modified syringe is not completely reproducible, this migration time varied by approximately 10% between runs. That the background signal remained relatively stable during the time of the assay suggests that the pressure applied by the modified syringe did not drop substantially during the assay period.
The flow rate obtained in this assay corresponds to a Reynold's number of <500, which is well within the range of laminar flow. As such, one would predict the product pool to remain well defined upon exiting the capillary. This was the case although Gaussian shaped peaks were not always observed. As such it was problematic to discern between two partially overlapping peaks and one due to a particularly active enzyme molecule. This could be largely alleviated by using low enzyme concentrations which reduces the likelihood of two enzyme molecules migrating in close proximity.
Capillary electrophoresis-based assays, in addition to measuring the activity distribution, are capable of multiple incubations of individual enzyme molecules at identical2,3 or different10 substrate concentrations as well as measurement of the electrophoretic mobility of the individual molecules11 or activation energy of catalysis.3 This is by virtue of the ability to separate the enzyme molecules from the product pools formed. This separation ability also allows the continuous flow-type assay which provides time resolution, allowing the determination of the change in catalytic rate of a given molecule over time.13 In instances where the substrate,15 or fluorescent impurities in the assay mixture,3 yield a high background signal, electrophoretic mobilization can be used to separate the product from these interfering compounds prior to detection. In this assay, no such separations are possible. The capillary is filled with substrate and enzyme and all the assay mixture components simultaneously and continuously migrate past the detector at identical rates. However, if the objective of the measurement is to solely determine the distribution of single molecule catalytic activities this method has distinct advantages. Not requiring electrophoresis, this system is simpler and less expensive to construct than the capillary electrophoresis-based protocols and is not prone to many of its technical challenges. Furthermore, this system is substantially less complex than the non-capillary electrophoresis-based protocols. Additionally, once initiated, the system can run on its own and without any operator input for more than 24 h.
Summary
Several systems have been developed which are capable of measuring the catalytic rate of individual enzyme molecules. In general, these systems employ equipment which can be somewhat complicated to construct and achieve proficiency with. Here a simple system was described which allows for the measurement of the activity of individual molecules of E. coli β-galactosidase. Although this system does not have the range of capability of other systems, such as multiple incubations of enzyme molecules or measurement of changes in activity over time, it is suitable for quantifying the heterogeneity with respect to catalytic activity of a population. In addition to its simplicity, this system was demonstrated to have the added advantage of being able to run on its own with absolutely no operator input for a period of longer than 24 h.Acknowledgements
This project was supported by a grant from the Natural Sciences and Engineering Research Council.References
- B. Rotman, Measurement of the activity of single molecules of β-D- galactosidase, Proc. Natl. Acad. Sci. U. S. A., 1961, 47, 1981–1991 CrossRef CAS.
- Q. Xue and E. S. Yeung, Differences in the chemical reactivity of individual molecules of an enzyme, Nature, 1995, 373, 681–683 CrossRef CAS.
- D. B. Craig, E. A. Arriaga, J. C. Y. Wong, H. Lu and N. J. Dovichi, Studies on single alkaline phosphatase molecules: reaction rate and activation energy of a reaction catalyzed by a single molecule and the effect of thermal denaturation— the death of an enzyme, J. Am. Chem. Soc., 1996, 118, 5245–5253 CrossRef CAS.
- W. Tan and E. S. Yeung, Monitoring the reactions of single enzyme molecules and single metal ions, Anal. Chem., 1997, 69, 4242–4248 CrossRef CAS.
- L. Edman, Z. Foldes-Papp, S. Wennmalm and R. Rigler, The fluctuating enzyme: A single molecule approach, Chem. Phys., 1999, 247, 11–22 CrossRef CAS.
- H. P. Lu, L. Xun and S. Xie, Single-molecule enzymatic dynamics, Science, 1999, 282, 1877–1882 CrossRef.
- H.-H. Gorris, D. M. Rissin and D. R. Walt, Stochastic inhibitor release and binding from single-enzyme molecules, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17680–17685 CrossRef CAS.
- D. M. Rissin, H.-H. Gorris and D. R. Walt, Distinct and long-lived activity states of single enzyme molecules, J. Am. Chem. Soc., 2008, 130, 5349–5353 CrossRef CAS.
- D. M. Rissin and D. R. Walt, Digital readout of target binding with attomolar detection limits via enzyme amplification in femtoliter arrays, J. Am. Chem. Soc., 2006, 128, 6286–6287 CrossRef CAS.
- D. B. Craig, A. M. Haslam, J. M. L. Coombs and E. R. Nichols, Kinetic studies on unmodified individual Escherichia coli β-galactosidase molecules in free solution, Biochem. Cell Biol., 2010, 88, 451–458 CrossRef CAS.
- E. R. Nichols and D. B. Craig, Measurement of the differences in electrophoretic mobilities of individual molecules of E. coli β-galactosidase provides insight into structural differences which underlie enzyme microheterogeneity, Electrophoresis, 2008, 29, 4257–4269 CrossRef CAS.
- K. Velonia, D. L. Flomenbom, S. Masuo, M. Cotlet, Y. Engelborghs, J. Hofkens, A. E. Rowan, J. Klafter, R. J. M. Nolte and F. C. deSchryver, Single-enzyme kinetics of CALB-catalyzed hydrolysis, Angew. Chem., Int. Ed., 2005, 44, 560–564 CrossRef CAS.
- D. B. Craig and E. R. Nichols, Continuous flow assay for the simultaneous measurement of the electrophoretic mobility, catalytic activity and its variation over time of individual molecules of Eschericia coli β-galactosidase, Electrophoresis, 2008, 29, 4298–4303 CrossRef CAS.
- J.-Y. Zhao, D.-Y. Chen and N. J. Dovichi, Low-cost laser-induced fluorescence detector for micellar capillary zone electrophoresis. Detection at the zeptomol level of tetramethylrhodamine thiocarbamyl amino acid derivatives, J. Chromatogr., A, 1992, 608, 117–120 CrossRef CAS.
- G. K. Shoemaker, D. H. Juers, J. M. L. Coombs, B. W. Matthews and D. B. Craig, Crystallization of β-galactosidase does not reduce the range of activity of individual molecules, Biochemistry, 2003, 42, 1707–1710 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |