Synthesis and characterization of a template-assembled synthetic U-quartet†
Benjamin Wei-Qiang
Hui
and
John C.
Sherman
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, Canada BC V6T 1Z1. E-mail: sherman@chem.ubc.ca; Fax: 1 604 822 2847; Tel: 1 604 822 2305
First published on 31st October 2011
Abstract
A lipophilic cavitand containing four triazole-linked uridine residues has been synthesized and characterized. Spectral evidence suggests a quartet arrangement of the uracil residues at both ambient and low temperatures. Treatment of the compound with Sr2+ yields a homodimeric complex as evidenced by NMR spectroscopy.
G-quartets and quadruplexes are supramolecular assemblies of guanine nucleobases held together by Hoogsteen hydrogen bonding.1 Their ubiquity in nucleic acids, especially within the telomeres of chromosomal DNA2 has linked them with such diseases as cancer, making them particularly attractive therapeutic targets.3 Organic templates have facilitated studies of these systems in vitro by reducing the entropic costs involved in their assembly, thereby enhancing thermodynamic stability and allowing more topological control.4 We recently demonstrated the self-assembly of a G-quartet on a cavitand template in chloroform.5 These template-assisted synthetic G-quartets (TASQs) displayed exceptional thermal stability even in the absence of central stabilizing metal cations.6
Unlike guanine, uracil (U) lacks the functionality needed to form Hoogsteen hydrogen bonds. Therefore, U-quartets are expected to be less stable than their G counterparts. Few reports exist of their occurrence. The first report of a U-quartet was in an RNA tetraplex of sequence r(UG4U)4 in the solution state.7NMR evidence suggested the N(3)–H(3)⋯O(4) hydrogen bond contacts of each of the uracil nucleobases (Fig. 1). Computational studies have correlated the same hydrogen bonding mode to an energy minimum.8 More recently, stability studies found that terminal U-quartets dramatically increased the melting temperature of a G-quadruplex.9 This might indicate a wider contribution of U-quartets to the stability of G-quadruplexes than previously thought. X-ray analyses of several crystalline adducts of uracil derivatives with heavy metal complexes have revealed unambiguously the existence of U-quartets in the solid state.10 Several RNA tetraplexes have also been crystallized in the presence of metal cations and display terminal U-quartets.11
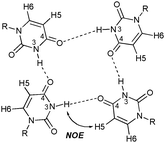 | ||
| Fig. 1 U-quartet showing the N(3)–H(3)⋯O(4) hydrogen bonds of each uracil residue. The H5 and H6 protons are also shown. The key NOE interaction between uracil's imino proton to H5 to suggest quartet formation is shown. | ||
So far, all documented examples of U-quartets place them at the termini of G-quadruplexes. To the best of our knowledge, no reports yet exist of isolated U-quartets. The study of such systems might potentially provide further insight on their stability and biological significance. Our cavitand templates therefore provide a unique opportunity for the assembly of an isolated U4 system in a lipophilic environment. Herein, we report the first synthesis and characterization of a lipophilic cavitand-uridine conjugate 1 (Fig. 2).
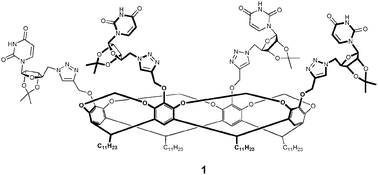
Towards the synthesis of conjugate 1, cavitand 2 was chosen as the template as the long aliphatic C11 chains at the lower rim of the cavitand architecture confer excellent lipophilicity. Both 25,12 and uridine derivative 313 were synthesized using known chemistry. The final step of the synthesis was accomplished via a copper-catalyzed azide-alkyne cycloaddition14 between 2 and 3, affording conjugate 1 in 47% yield (Scheme 1). The 1H NMR spectrum of 1 was assigned by 1H-1H COSY in both CDCl3 and DMSO-d6.
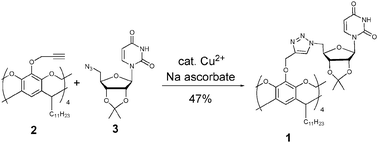 | ||
| Scheme 1 Synthesis of conjugate 1viaCu-catalyzed azide-alkyne cycloaddition. | ||
Corey–Pauling–Koltun (CPK) modeling of 1 demonstrated the possibility of a planar U-quartet lying atop the cavitand template with the expected N(3)–H(3)⋯O(4) hydrogen bond pattern between the uracil moieties. 2D NOESY of 1 at room temperature reveals a cross-peak between the imino NH proton and H5, the key correlation that has been used to identify U-quartets (Fig. 3a).7 The low intensity of this cross-peak suggests a rapid exchange between the ordered (U-quartet) and a random (non U-quartet) state. We surmised that lowering the temperature might slow down the exchange sufficiently to obtain a more intense imino NH/H5 cross-peak. This was observed at −20 °C (Fig. 3b). It is also interesting to note that the imino NH and H5 signals show broadening at this temperature with respect to ambient conditions. This also supports the notion of a slowed exchange.
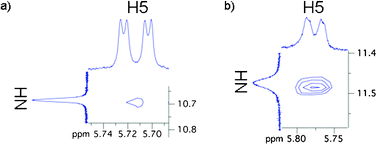 | ||
| Fig. 3 Portions of NOESY spectra of 1 in CDCl3 acquired at 400 MHz indicative of a U-quartet (a) at room temperature, (b) at −20 °C. | ||
The CPK model also showed that both syn and anti conformations of the uracil residues are feasible (Fig. 4a). A strong NOE cross-peak observed between H6 of uracil and H1′ of the ribose sugar revealed the system to be in the syn configuration (Fig. 4b).
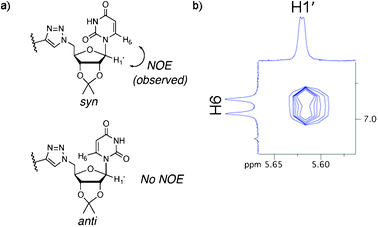 | ||
| Fig. 4 (a) Syn and anti conformations of uracil in conjugate 1 and (b) the H6–H1′ NOE cross-peak indicative of the syn conformation. | ||
Circular dichroism (CD) spectroscopy of a solution of 1 in chloroform revealed a signal with λmax ∼245 nm (Fig. 5). It was not possible to obtain a spectral profile below 230 nm due to the absorption limit of chloroform. In methanol as solvent, a shift of λmax to ∼255 nm was observed. Solvatochromic shifting was ruled out by the use of a modified conjugate 4 (Fig. 6) in which the imino positions of the uracil bases are methylated, negating the possibility of quartet formation: Spectra of 4 obtained in both chloroform and methanol were indistinguishable with λmax ∼254 nm. It is noteworthy that the spectral profile of 1 in methanol is similar to that of 4 in either solvent. This suggests a non-quartet state of 1 in methanol as it disrupts H-bonds.
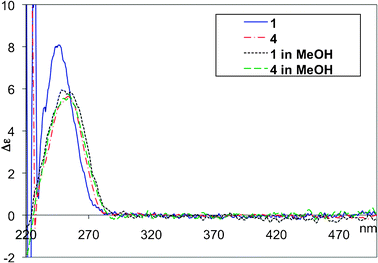 | ||
| Fig. 5 CD spectra of 0.1 mM solutions of 1 and 4 in CHCl3 and MeOH. | ||
 | ||
| Fig. 6 Side-on view of modified conjugate 4. | ||
We next investigated the behaviour of 1 in the presence of Sr2+. It is well known that metal cations are able to bind and stabilize G-quartets and quadruplexes. The same premise was envisioned for our system. Sr2+ was chosen because it bound most strongly to the TASQs in our previous work,6 and other cations (Na+ and K+) were found not to be taken up by 1. To this end, we performed metal cation extraction experiments on 1 with Sr2+ by means of its picrate salt Sr(pic)2. The salt was prepared according to a literature procedure.15 A solution of 1 in chloroform was stirred with an excess of Sr(pic)2 for 7 days, after which the solution was filtered and subjected to CD spectroscopy. A signal was observed from 300–450 nm and attributed to the picrate anions16 being held in a chiral environment (Fig. 7). This suggests Sr2+ is sequestered by the conjugate in chloroform and a conjugate-metal picrate complex has assembled (1·Sr2+). λmax for this species was observed at ∼245 nm, identical to 1 in chloroform. On switching the solvent to methanol, the picrate signal is quenched and a bathochromic shift occurs with the new λmax at ∼256 nm, identical to 1 in methanol, showing that it has a destabilizing effect on the formed complex.
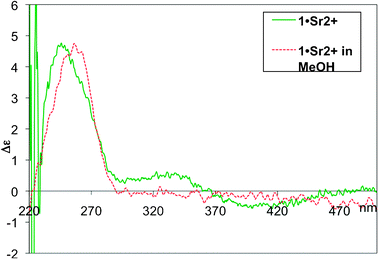 | ||
| Fig. 7 CD spectra of 0.1 mM solutions of 1·Sr2+ in CHCl3 and MeOH. | ||
The 1H NMR spectrum (see Supporting Information) of 1·Sr2+ shows the picrate aromatic proton signal at 8.7 ppm, which on integration reveals a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio of 1 to Sr(pic)2 suggesting a symmetric dimeric complex. Diffusion-ordered spectroscopy (DOSY) was performed on 1, 1·Sr2+ and 4, and the measured diffusion coefficients (D) of the H1′ on the three species are 3.71 × 10−10, 3.11 × 10−10 and 3.67 × 10−10 m2 s−1 respectively. D values of 1 and 4 are within 1% of each other, indicating the oligomeric state of these two species to be identical. In contrast, the diffusion coefficient of 1·Sr2+ is only 84% that of 1. This is in close agreement with the theoretical value computed for dimeric with respect to monomeric spherical molecular systems.17
:1 stoichiometric ratio of 1 to Sr(pic)2 suggesting a symmetric dimeric complex. Diffusion-ordered spectroscopy (DOSY) was performed on 1, 1·Sr2+ and 4, and the measured diffusion coefficients (D) of the H1′ on the three species are 3.71 × 10−10, 3.11 × 10−10 and 3.67 × 10−10 m2 s−1 respectively. D values of 1 and 4 are within 1% of each other, indicating the oligomeric state of these two species to be identical. In contrast, the diffusion coefficient of 1·Sr2+ is only 84% that of 1. This is in close agreement with the theoretical value computed for dimeric with respect to monomeric spherical molecular systems.17
In summary, we have synthesized and characterized the first example of a uridine-cavitand conjugate 1. NMR evidence suggests the presence of a U-quartet assembly both at ambient and low temperature. To the best of our knowledge, this is the first example of an isolated U-quartet in a lipophilic environment. In the presence of Sr2+, a homodimeric complex involving two molecules of 1 and one of Sr(pic)2 is formed, as evidenced by 1H-NMR, DOSY and CD spectroscopy. Work is currently in-progress to create uracil-based monomeric quadruplexes.
Notes and references
- M. Gellert, M. N. Lipsett and D. R. Davies, Proc. Natl. Acad. Sci. U. S. A., 1962, 48, 2013 CrossRef CAS.
- J. R. Williamson, Annu. Rev. Biophys. Biomol. Struct., 1994, 23, 703 CrossRef CAS.
- (a) S. Balasubramanian, L. H. Hurley and S. Neidle, Nat. Rev. Drug Discovery, 2011, 10, 261 CrossRef CAS; (b) S. Neidle, FEBS J., 2010, 277, 1118 CrossRef CAS; (c) A. T. Phan, FEBS J., 2010, 277, 1107 CrossRef CAS; (d) S. Neidle, Curr. Opin. Struct. Biol., 2009, 19, 239 CrossRef CAS; (e) S. Balasubramanian and S. Neidle, Curr. Opin. Struct. Biol., 2009, 13, 345 CAS; (f) A. Arola and R. Vilar, Curr. Top. Med. Chem., 2008, 8, 1405 CrossRef CAS; (g) T.-M. Ou, Y.-J. Lu, J.-H. Tan, Z. S. Huang, K.-Y. Wong and L.-Q. Gu, ChemMedChem, 2008, 3, 690 CrossRef CAS; (h) D. J. Patel, A. T. Phan and V. Kuryavyi, Nucleic Acids Res., 2007, 35, 7429 CrossRef CAS.
- (a) P. Murat, R. Bonnet, A. Van der Heyden, N. Spinelli, P. Labbe, D. Monchaud, M.-P. Teulade-Fichou, P. Dumy and E. Defrancq, Chem. Eur. J., 2010, 16, 6106 CAS; (b) P. Murat, D. Cressend, N. Spinelli, A. Van der Heyden, P. Labbe, P. Dumy and E. Defranq, ChemBioChem, 2008, 9, 2588 CrossRef CAS; (c) G. Oliviero, N. Borbone, A. Galeone, M. Varra, G. Piccialli and L. Mayol, Tetrahedron Lett., 2004, 45, 4869 CrossRef CAS; (d) V. Sidorov, F. W. Kotch, M. El-Kouedi and J. T. Davis, Chem. Commun., 2000, 2369 RSC.
- N. Mehran and J. C. Sherman, Angew. Chem., Int. Ed., 2008, 47, 4900 CrossRef.
- N. Mehran and J. C. Sherman, J. Org. Chem., 2009, 74, 5211 CrossRef.
- C. Cheong and P. B. Moore, Biochemistry, 1992, 31, 8406 CrossRef CAS.
- (a) X. Zhao and F. Meng, THEOCHEM, 2006, 770, 157 CrossRef CAS; (b) J. Gu and J. Leszczynski, J. Phys. Chem. A, 2001, 105, 10366 CrossRef CAS; (c) M. Meyer, T. Steinke, M. Brandl and J. Suhnel, J. Comput. Chem., 2001, 22, 109 CrossRef CAS.
- X. Yan, T. Ishizuka, T. Kimura and M. Komiyama, J. Am. Chem. Soc., 2010, 132, 7231 CrossRef.
- (a) M. Barcelo-Oliver, C. Estarellas, A. Terron, A. Garcia-Raso and A. Frontera, Chem. Commun., 2011, 47, 4646 RSC; (b) H. Witkowski, E. Freisinger and B. Lippert, Chem. Commun., 1997, 1315 RSC.
- (a) B. Pan, Y. Xiong, K. Shi and M. Sundaralingam, Structure, 2003, 11, 825 CrossRef CAS; (b) J. Deng, Y. Xiong and M. Sundaralingam, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13665 CrossRef CAS.
- D. J. Cram, R. Jaeger and K. Deshayes, J. Am. Chem. Soc., 1993, 115, 10111 CrossRef CAS.
- F. Liu and D. J. Austin, J. Org. Chem., 2001, 66, 8643 CrossRef CAS.
- V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- K. H. Wong and H. L. Ng, J. Coord. Chem., 1981, 11, 49 CrossRef CAS.
- (a) U. Olsher, H. Feinberg, F. Frolow and G. Shoham, Pure Appl. Chem., 1996, 68, 1195 CrossRef CAS; (b) W. R. Gilkerson and A. M. Roberts, J. Am. Chem. Soc., 1980, 102, 5181 CrossRef CAS; (c) M. Bourgoin, K. H. Wong, J. Y. Hui and J. Smid, J. Am. Chem. Soc., 1975, 97, 3462 CrossRef CAS; (d) K. H. Wong, M. Bourgoin and J. Smid, J. Chem. Soc., Chem. Commun., 1974, 715 RSC.
- A. R. Waldeck, P. W. Kuchel, A. J. Lennon and B. E. Chapman, Prog. Nucl. Magn. Reson. Spectrosc., 1997, 30, 39 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra. See DOI: 10.1039/c1cc15608j |
| This journal is © The Royal Society of Chemistry 2012 |