Ultra-trace determination of plutonium in urine samples using a compact accelerator mass spectrometry system operating at 300 kV
Xiongxin
Dai
*a,
Marcus
Christl
b,
Sheila
Kramer-Tremblay
a and
Hans-Arno
Synal
b
aChalk River Laboratories, Atomic Energy of Canada Limited, Chalk River, Ontario K0J 1J0, Canada. E-mail: daix@aecl.ca; Tel: +1 613 584 3311 ext. 46128
bLaboratory of Ion Beam Physics, ETH-Zurich, CH-8093, Zurich, Switzerland
First published on 15th November 2011
Abstract
Accelerator mass spectrometry (AMS) is a very sensitive and robust technique for analysis of long-lived radionuclides. Employment of the AMS technique can reduce the demands on sample preparation chemistry, due to its high rejection of interferences and low susceptibility to sample matrix. This is particularly of interest for ultra-trace determination of 239Pu in bioassay and environmental samples, as other mass spectrometric methods such as inductively coupled plasma mass spectrometry (ICP-MS) can suffer from isobaric mass interferences by the presence of uranium in the sample. A rapid sample preparation method for analysis of Pu at femtogram levels in large volume urine samples is described. Using the compact ETH AMS Tandy facility operating at ∼300 kV, the method was validated by analysing urine samples spiked with known amounts of 239/240/241Pu ranging from 1 to 30 fg. The detection limits for the method were estimated to be 0.38 fg for 239Pu, 0.40 fg for 240Pu and 0.08 fg for 241Pu in 1400 mL of urine.
Introduction
Plutonium is present in varying concentrations in environmental and biological samples as a consequence of activities related to nuclear weapons testing, nuclear waste recycling, and accidental or authorised discharges from nuclear power plants. Plutonium is considered to be the most radiotoxic substance, in particular, when it gets into the human body through inhalation of insoluble dust or aerosol. Occupational exposure to Pu usually comes from inhalation.1,2 The most commonly used methods to assess internal contamination to Pu are in vitro measurements of its concentration in body fluids, especially in urine samples. As Canadian Nuclear Safety Commission (CNSC) regulations require the ability to report all occupational exposures of 1 mSv or greater committed effective dose, there is an urgent need for ultra-sensitive urine bioassay methods for dose assessment of Pu exposures for nuclear energy workers at the Canadian nuclear utilities. The International Commission on Radiological Protection (ICRP) has also recommended an annual dose limit of 1 mSv for the general public.3 To meet these regulatory requirements, a highly sensitive and accurate method capable of measuring the 239/240/241Pu isotopes at a concentration of ∼1 fg L−1 in urine samples is required.Although measurement of Pu isotopes has been routinely undertaken by a number of techniques such as alpha spectrometry4,5 and ICP-MS,6,7 it is very difficult to accurately and precisely measure Pu isotopes by these techniques at fg levels in a large volume of urine sample. The minimum detectable activity by alpha spectrometry (AS) is around 0.1 mBq (∼50 fg) for 239Pu, which does not provide sufficient sensitivity to detect the Pu at the low fg levels required for occupational urine bioassay samples. Another disadvantage is that AS cannot provide simultaneous measurements of the 239Pu, 240Pu and 241Pu isotopes, due to very similar alpha energy emissions between 239Pu and 240Pu and very low alpha branching ratio (2.45 × 10−5) for 241Pu. In spite of the fact that recent developments of the ICP-MS techniques have pushed the detection limit for analysis of 239Pu down to sub-fg level,7–11 employment of the ICP-MS techniques for routine Pu bioassay at low fg L−1 concentration in urine is still very challenging. The presence of high content of uranium in urine could cause significant interferences with 239Pu by ICP-MS due to uranium hydride (238UH+) formation and peak tailing from neighbouring 238U+ mass.12,13
Other techniques that have been used in the determination of fg quantities of 239Pu in urine include fission track analysis (FTA),14,15 thermal ionization mass spectrometry (TIMS)16–19 and AMS.19–23 The main disadvantage of FTA method is its large uncertainty in the results caused by interference from 235U in the sample which can produce indistinguishable fission tracks for 239Pu analysis. TIMS has been successfully applied to ultra-sensitive urine bioassay, but the sample analysis throughput is quite limited as preparation of the loading filament requires very tedious and time-consuming chemical separation of the sample matrix elements.
The AMS technique has also been demonstrated for the precise and accurate determination of Pu at sub-fg level in urine bioassay samples.20–23AMS is considered to be the most sensitive and robust technique because of its high rejection of molecular isobaric interferences and low susceptibility to matrix effects, which allow simplification of sample preparation chemistry. As a result of its high sensitivity and easy chemical separation, there is a good potential of high sample analysis throughput for routine Pu bioassay by AMS. However, lack of availability of accelerators and high costs to operate the complicated AMS system have limited extensive use of this technique. Recent developments at ETH Zurich demonstrated the applicability of a new generation compact AMS system for actinide analysis at low fg levels.24,25 This compact system, working with a terminal voltage of 300 kV, is much simpler and more cost efficient. This makes low energy AMS a very competitive and cost effective technique compared to conventional mass spectrometers.
Based on the previous development of a rapid emergency bioassay method for actinides,26 a new radiochemical separation method followed by AMS analysis was developed for isotopic Pu analysis in 24 hour urine bioassay samples. Using the ETH compact AMS, the method was validated with spiked urine samples.
Experimental
Reagents and standards
The anion exchange resin was AGMP-1M (Cl− form, 100–200 mesh), purchased from Bio-Rad Laboratories Canada Ltd. (Mississauga, Ontario, Canada). The deionized water (>18 MΩ cm−1) was obtained from a Millipore Direct-Q5 ultrapure water system. All the other chemicals were supplied by Sigma-Aldrich Canada. Most of these chemicals were trace metal grade or above, except for titanium oxychloride, which was of analytical grade. Radiochemical isotope standards were purchased from the National Institute of Standards and Technology (NIST, Gaithersburg, MD, USA), including 239Pu (NIST 4330B), 241Pu (NIST 4340B) and 242Pu (NIST 4334G). Based on the TIMS analysis, both the 239Pu and 242Pu standards were found to be sufficiently pure with no measurable interferences from other Pu isotopes. However, significant impurities of 239Pu and 240Pu were found in the 241Pu standard. The 239/240Pu concentrations in the 241Pu standard were successfully calibrated by TIMS after anion exchange purification to remove isobaric interference from the 241Am progeny to 241Pu. Thus, the 239/240Pu measurements in the samples spiked with the 241Pu standard can also be used for comparison to evaluate the performance of the present procedure.Samples
To validate the Pu bioassay procedure, the spike samples were prepared by adding a known amount of the 239Pu or 241Pu standard solutions into 1400 mL of synthetic urine or pooled urine. The synthetic urine was prepared according to the recipe published by McCurdy et al.19 To correct the procedural backgrounds, the reagent blanks were prepared using 500 mL of deionised water as a sample, which was acidified with 50 mL of concentrated HNO3 and was processed following through the entire procedure. The urine blank samples were also prepared as 1400 mL of synthetic or pooled urine for subtraction of the Pu backgrounds in urine from the spiked samples.Sample preparation procedure
A flow diagram of the sample preparation procedure is shown in Fig. 1. 1400 mL of urine sample was transferred into a glass beaker and was acidified with 50 mL concentrated nitric acid. A known quantity of 242Pu tracer (∼5 pg of 242Pu) was then added to monitor the chemical recovery. The sample was well mixed at low heating temperature (<80 °C) for about one hour and cooled down to room temperature.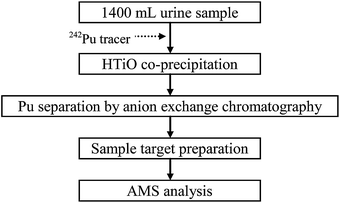 | ||
| Fig. 1 Flow diagram of the Pu urinalysis procedure. | ||
In the hydrous titanium oxide (HTiO) co-precipitation step, a suitable amount of TiOCl2 solution was added. After stirring, the sample was neutralized to pH ∼7 with concentrated NH4OH, and plutonium was co-precipitated with the formation of HTiO from the urine matrix. The sample was stirred for >15 minutes and transferred to a centrifuge bottle. After centrifugation at 3500 rpm for 3 minutes, the supernatant solution was decanted. The precipitate was then dissolved and transferred to a Teflon beaker with ∼10 mL concentrated HNO3. The sample was heated to boiling with the addition of 0.5 mL of H2O2 to decompose the residual organics. The sample was then neutralized with NH4OH, and the supernatant solution was decanted after centrifugation. The HTiO precipitate was rinsed with water and centrifuged to remove residual salts. In the subsequent step, the precipitate was dissolved in 8 M HNO3 with the addition of 0.5 mL of H2O2 for anion exchange purification.
The anion exchange column, containing 2 mL of AGMP-1M resin, was pre-conditioned with 10 mL of water followed by 8 M HNO3 before use. The sample was passed through the column at ∼1 mL min−1. After being rinsed with 8 M HNO3, any thorium extracted onto the resin was removed with 5 mL of concentrated HCl. Plutonium was eluted with 10 mL of 0.2 M HNO3 + 0.05 M HF.
The AMS target was prepared by either iron hydroxide or titanium hydroxide co-precipitation methods. To do this, ∼1 mg of Fe or Ti in the stock standard solutions was added, and the sample was neutralized with NH4OH to pH > 9 to co-precipitate the Pu isotopes. The supernatant solution was decanted after centrifugation, and the hydroxide precipitate was dried under an infrared lamp. The dried sample was baked in a furnace for 2–3 hours at 650 °C. It was finally mixed with 4–5 mg niobium powder and pressed into a Ti target holder for the AMS measurement.
AMS analysis
The AMS measurements were performed with the compact (0.6 MV) AMS system TANDY at ETH Zurich.27 Since the AMS set-up for actinide measurements is described in detail elsewhere,24,25,28 only the main points and the most recent developments are summarized here. Negatively charged PuO ions are extracted from the Cs-sputter ion source and injected into the accelerator running at a terminal voltage of about 300 kV. At the terminal, helium is used as a stripper gas to break up the injected molecules and to generate positively charged Pu ions. On the high energy side, Pu3+ ions are selected. Recently, it has been demonstrated that the transmission for the actinides, Th and U through the accelerator (into the 3+ charge state), increased from about 12% when using Ar-stripping to almost 40% when using He-stripping.29 This implies that the sensitivity for Pu-detection with the compact ETH AMS system Tandy has increased by a factor of three compared to the previous set-up. The final ion identification is made with a dedicated low noise gas ionization detector30 equipped with a 30 nm thick SiN entrance window.31 The Pu isotopic ratios of interest (i.e., 239Pu/242Pu, 240Pu/242Pu, and 241Pu/242Pu) were determined by a sequential measurement. Switching between the different Pu-isotopes typically takes two seconds (1 s for switching plus 1 s of idle time to ensure stable operation conditions). To enable fast isotope switching, only electrostatic devices (no magnetic fields) are changed (including the pulsing system on the low energy side, the acceleration voltage, and the electrostatic analyser on the high energy side). The measurement time for each isotope can be adjusted individually for each set of samples according to the expected counting rate. For this study, 239Pu, 240Pu, and 241Pu were measured for 25 s each, while the 242Pu tracer was counted for 8 s. In addition, for each sample, the background counting rate on amu (atomic mass unit) 239 was monitored for 8 s by injecting 238U16O− into the accelerator while leaving the high energy side tuned to amu 239.To summarize, the measurement of one sequence as described above takes about 100 s. This sequence was repeated four times before measuring the next sample. The isotopic ratios of interest were calculated from the average counting rates acquired during the repeated sequential measurements. The whole measurement procedure as described above was repeated 8–12 times for each set of samples. The final Pu isotopic ratios were calculated from the error weighted mean of all single measurements. In total, each Pu sample was measured between 60 and 90 minutes on average.
Results and discussion
The mass of the analyte Pu isotopes (including 239Pu, 240Pu and 241Pu) present in the samples was calculated from the measured ratio of the respective Pu isotope to the 242Pu tracer (e.g., 239Pu/242Pu, see above) by reference to the known amount of the tracer added. The average counting rate derived from the 242Pu tracer depends on the actual operating conditions of the ion source as well as the chemical recovery of the sample preparation procedure. The blanks and urine samples presented in this study typically yielded between 40 and 300 counts s−1 pg−1 at amu 242. Background corrections (e.g., for 238U tailing on amu 239) were negligible for all samples. The cross-talk in the ion source (i.e., the potential interference caused when the sputtered material re-deposits on the sample wheel, and thus, contaminates neighbouring samples) was on the order of 10−6 for both sets of samples.Procedural blank and detection limit
Two sets of samples, including reagent (procedural) blanks, urine blanks, and spike samples, were prepared to evaluate the performance of the method. The first set of samples were prepared at the Chalk River Laboratories (CRL) in December 2010 and analysed at the ETH in February 2011; while the second set of samples were prepared in April 2011 and analysed in May 2011. The results for the blank samples are listed in Table 1.| Sample | Description | 239Pu/fg | 240Pu/fg | 241Pu/fg |
|---|---|---|---|---|
| The first set of samples | ||||
| T1-8 | Reagent blank | 1.62 ± 0.45 | 0.84 ± 0.30 | 0.84 ± 0.30 |
| T1-9 | Reagent blank | 1.95 ± 0.50 | 0.97 ± 0.43 | 0.97 ± 0.43 |
| T1-6 | Synthetic urine blank | 0.92 ± 0.24 | 0.93 ± 0.30 | 0.93 ± 0.30 |
| T1-7 | Synthetic urine blank | 2.18 ± 0.53 | 1.36 ± 0.43 | 1.36 ± 0.43 |
| T1-14 | Pooled urine blank | 1.93 ± 0.28 | 1.40 ± 0.23 | 1.40 ± 0.23 |
| The second set of samples | ||||
| T2-7 | Reagent blank | 0.57 ± 0.05 | 0.27 ± 0.04 | 0.05 ± 0.04 |
| T2-8 | Reagent blank | 0.30 ± 0.04 | 0.13 ± 0.03 | <0.01 |
| T2-9 | Reagent blank | 0.44 ± 0.11 | 0.40 ± 0.10 | <0.02 |
| T2-11 | Pooled urine blank | 0.46 ± 0.07 | 0.15 ± 0.05 | 0.07 ± 0.05 |
| T2-12 | Pooled urine blank | 0.59 ± 0.24 | 0.36 ± 0.19 | <0.04 |
Higher Pu backgrounds were found in the first set of samples due to possible cross-contamination introduced during the AMS target preparation step, as several samples containing much higher Pu content (at picogram levels) were dried together with this set of samples under the same infrared lamp. The laboratory cross-contamination significantly decreased for the second set of samples by cleaning the lamp and careful control of the sample drying step; thus, lower Pu backgrounds (especially for 241Pu) were achieved. In addition, no apparent difference was seen between the reagent and urine blank samples, suggesting that the Pu backgrounds are dominated by the reagents used in the procedure.
Based on the measurements of all the reagent and urine blanks for the second set of samples, the average procedural backgrounds were found to be 0.47 ± 0.12 fg for 239Pu, 0.26 ± 0.12 fg for 240Pu and 0.038 ± 0.024 fg for 241Pu. Assuming a standardized normal distribution for the procedural blanks, the detection limit (Ld) can be calculated by:32
| Ld = 2 × 1.645 × σ0 |
Performance tests for spike samples
The results of the urine samples spiked with known amounts of 241Pu and 239Pu standard solutions are summarized in Table 2 and Table 3, respectively. The measured values have been blank corrected with ±1σ uncertainties. Measurements for the 239/240/241Pu isotopes ranged from ∼1 to 30 fg per 1400 mL of urine sample, agreeing very well with the expected values. As a result of higher blanks found in the first set of samples, elevated detection limits of <1.6 fg 239Pu and <1.3 fg 240Pu for the samples T1-12, T1-13 and T1-16 are reported in Table 2.| Sample | Description | 241Pu | 239Pu | 240Pu | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Added/fg | Measured/fg | Deviation | Added/fg | Measured/fg | Deviation | Added/fg | Measured/fg | Deviation | ||
| The first set of samples | ||||||||||
| T1-12 | Synthetic urine spike | 5.78 | 6.1 ± 1.2 | 6.0% | 0.3 | <1.6 | — | 1.2 | <1.3 | — |
| T1-13 | Synthetic urine spike | 27.3 | 27.5 ± 1.3 | 1.0% | 1.5 | <1.6 | — | 5.6 | 5.2 ± 0.6 | −5.7% |
| T1-16 | Pooled urine spike | 11.4 | 12.4 ± 1.2 | 9.5% | 0.6 | <1.6 | — | 2.3 | 2.8 ± 0.8 | 19.1% |
| The second set of samples | ||||||||||
| T2-15 | Pooled urine spike | 20.6 | 20.0 ± 0.3 | −2.9% | 1.1 | 1.2 ± 0.1 | 6.4% | 4.2 | 4.7 ± 0.2 | 11.9% |
| T2-18 | Pooled urine spike | 16.9 | 16.2 ± 0.4 | −3.8% | 0.9 | 0.7 ± 0.3 | −27.9% | 3.4 | 3.5 ± 0.3 | 1.6% |
| Sample | Description | 239Pu | ||
|---|---|---|---|---|
| Added/fg | Measured/fg | Deviation | ||
| The first set of samples | ||||
| T1-10 | Synthetic urine spike | 3.59 | 2.9 ± 0.5 | −19.7% |
| T1-11 | Synthetic urine spike | 30.1 | 31.4 ± 1.1 | 4.4% |
| T1-15 | Pooled urine spike | 13.7 | 13.8 ± 0.9 | 0.9% |
| The second set of samples | ||||
| T2-13 | Pooled urine spike | 18.6 | 19.6 ± 1.3 | 5.1% |
| T2-14 | Pooled urine spike | 18.4 | 20.2 ± 0.7 | 9.8% |
| T2-16 | Pooled urine spike | 16.0 | 16.7 ± 0.8 | 4.5% |
| T2-17 | Pooled urine spike | 15.4 | 14.3 ± 0.5 | −7.3% |
As shown in Table 4, relative bias (Br) and relative precision (SB) are calculated as defined by the guidance provided in ANSI/HPS N13.30.33 The validation tests for all the analyte Pu isotopes passed the ANSI/HPS N13.30 performance criteria for both relative bias (−25% to +50%) and precision (±40%) by a very substantial margin, demonstrating the excellent accuracy and precision of Pu isotopic analysis with the present method.
| 239Pu | 240Pu | 241Pu | |
|---|---|---|---|
| Relative bias (Br) | −0.3% | 6.7% | 2.0% |
| Relative precision (SB) | 10.0% | 10.6% | 5.7% |
Conclusions
AMS is an extremely sensitive and very reliable technique for isotopic analysis of plutonium at low femtogram ranges. In this study, a new ultra-trace bioassay method for the determination of fg amounts of 239Pu, 240Pu and 241Pu in a large volume urine sample has been developed. The performance tests of the method were carried out, and the Pu isotopes were analysed using the compact ETH AMS system operating at 300 kV. The method is relatively simple, fast and robust, with a sensitivity of <0.5 fg for Pu. The method validation tests using spiked urine samples showed excellent agreement between the measured and the expected values for Pu at 1–30 fg in 1400 mL of urine. The results demonstrate the feasibility of the AMS technique using a compact, low energy AMS system for routine isotopic Pu analysis at ultra-trace levels in urine samples, to meet the regulatory requirement for a reportable dose of 1 mSv for occupational Pu exposures in the Canadian nuclear facilities.Acknowledgements
The authors wish to thank Dr Nicholas Priest (the AECL Chalk River Laboratories), Dr Xiaolin Hou (Risø National Laboratory for Sustainable Energy, Technical University of Denmark) and Dr Vasily Alfimov (ETH Zurich) for helpful discussions and suggestions. Further acknowledgements go to Johannes Lachner (ETH Zurich) for supporting the AMS measurements and for discussions and to Michael Rüttimann for carefully preparing the Pu-AMS targets at ETH Zurich. This study was partially funded by Atomic Energy of Canada Limited.References
- J. H. Diel and J. A. Mewhinney, Health Phys., 1983, 44, 133–145 CrossRef.
- E. H. Carbaugh, D. E. Bihl and M. J. Sula, Radiat. Prot. Dosim., 1991, 38, 99–104 CAS.
- International Commission on Radiological Protection (ICRP), The 2007 Recommendations of the International Commission on Radiological Protection, ICRP Publication 103, JAICRP 37, 2007 Search PubMed.
- D. Arginelli, G. Berton, S. Bortoluzzi, G. Canuto, F. Groppi, M. Montalto, M. Nocente, S. Ridone and M. Vegro, J. Radioanal. Nucl. Chem., 2008, 277, 65–71 CrossRef CAS.
- A. Thakkar, J. Radioanal. Nucl. Chem., 2002, 252, 215–218 CrossRef CAS.
- E. J. Wyse and D. R. Fisher, Radiat. Prot. Dosim., 1994, 55, 199–206 CAS.
- M. V. Zoriy, C. Pickhardt, P. Ostapczuk, R. Hille and J. S. Becker, Int. J. Mass Spectrom., 2004, 232, 217–224 CrossRef CAS.
- J. Zheng and M. Yamada, Talanta, 2006, 69, 1246–1253 CrossRef CAS.
- M. Liezers, S. A. Lehn, K. B. Olsen, O. T. Farmer, III and D. C. Duckworth, J. Radioanal. Nucl. Chem., 2009, 282, 299–304 CrossRef CAS.
- D. Schaumloffel, P. Giusti, M. V. Zoriy, C. Pickhardt, J. Szpunar, R. Yobinski and J. S. Becker, J. Anal. At. Spectrom., 2005, 20, 17–21 RSC.
- P. Lindahl, M. Keith-Roach, P. Worsfold, M. S. Choi, H. S. Shin and S. H. Lee, Anal. Chim. Acta, 2010, 671, 61–69 CrossRef CAS.
- F. Pointurier, P. Hemet and A. Hubert, J. Anal. At. Spectrom., 2008, 23, 94–102 RSC.
- F. Pointurier, A. Hubert, A. L. Faure, P. Hemet and A. C. Pottin, J. Anal. At. Spectrom., 2011, 26, 1474–1480 RSC.
- L. C. Sun, A. R. Moorthy, E. Kaplan, J. W. Baum and C. B. Meinhold, Appl. Radiat. Isot., 1995, 46, 1259–1269 CrossRef CAS.
- L. Johansson and E. Holm, Nucl. Instrum. Methods Phys. Res., Sect. A, 1996, 376, 242–247 CrossRef CAS.
- N. L. Elliot, G. A. Bickel, S. H. Linauskas and L. M. Paterson, J. Radioanal. Nucl. Chem., 2006, 267, 637–650 CrossRef CAS.
- S. P. LaMont, C. R. Shick, P. Cable-Dunlap, D. J. Fauth and T. R. LaBone, J. Radioanal. Nucl. Chem., 2005, 263, 477–481 CAS.
- D. Lewis, G. Miller, C. J. Duffy, D. W. Efurd, W. C. Inkret and S. E. Wagner, J. Radioanal. Nucl. Chem., 2001, 249, 115–120 CrossRef CAS.
- A. McCurdy, Z. Lin, K. G. W. Inn, R. Bell, III, S. Wagner, D. W. Efurd, R. Steiner, C. Duffy, T. F. Hamilton, T. A. Brown and A. A. Marchetti, J. Radioanal. Nucl. Chem., 2005, 263, 447–455 Search PubMed.
- A. A. Marchetti, T. A. Brown, C. C. Cox, T. F. Hamilton and R. E. Martinelli, J. Radioanal. Nucl. Chem., 2005, 263, 483–487 CAS.
- N. D. Priest, G. M. Pich, L. K. Fifield and R. G. Cresswell, Radiat. Res., 1999, 152, S16–S18 CrossRef CAS.
- H. Hernández-Mendoza, E. Chamizo, A. Yllera, M. García-León and A. Delgado, Nucl. Instrum. Methods Phys. Res., Sect. B, 2010, 268, 1331–1333 CrossRef.
- H. Hernández-Mendoza, E. Chamizo, A. Yllera, M. García-León and A. Delgado, J. Anal. At. Spectrom., 2010, 25, 1410–1415 RSC.
- L. Wacker, E. Chamizo, L. K. Fifield, M. Stocker, M. Suter and H. A. Synal., Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 240, 452–457 CrossRef CAS.
- M. Christl, L. Wacker, J. Lippold, H. A. Synal and M. Suter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 262, 379–384 CrossRef CAS.
- X. Dai and S. Kramer-Tremblay, Health Phys., 2011, 101, 144–147 CrossRef CAS.
- M. Stocker, M. Döbeli, M. Grajcar, M. Suter, H. A. Synal and L. Wacker, Nucl. Instrum. Methods Phys. Res., Sect. B, 2005, 240, 483–489 CrossRef CAS.
- L. K. Fifield, H. A. Synal and M. Suter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 223–24, 802–806 CrossRef.
- C. Vockenhuber, M. Christl, C. Hofmann, J. Lachner, A. M. Müller and H. A. Synal, Nucl. Instrum. Methods Phys. Res., Sect. B, 2011 DOI:10.1016/j.nimb.2011.04.026.
- M. Suter, M. Döbeli, M. Grajcar, A. Müller, M. Stocker, G. Y. Sun, H. A. Synal and L. Wacker, Nucl. Instrum. Methods Phys. Res., Sect. B, 2007, 259, 165–172 CrossRef CAS.
- M. Döbeli, C. Kottler, M. Stocker, S. Weinmann, H. A. Synal, M. Grajcar and M. Suter, Nucl. Instrum. Methods Phys. Res., Sect. B, 2004, 219–220, 415–419 CrossRef.
- L. A. Currie, Anal. Chim. Acta, 1999, 391, 127–134 CrossRef CAS.
- Health Physics Society (HPS), Performance Criteria for Radiobioassay: an American National Standard, ANSI/HPS N13.30, American National Standard Institute, New York, 1996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |