Efficient and continuous monoacylation with superior selectivity of symmetrical diamines in microreactors†
Ram Awatar
Maurya
a,
Phan Huy
Hoang
a and
Dong-Pyo
Kim
*ab
aNational Creative Research Centre of Applied Microfluidic Chemistry, Chungnam National University, Daejeon, 305-764, South Korea
bGraduate School of Analytical Science and Technology, Chungnam National University, Daejeon, 305-764, South Korea. E-mail: dpkim@cnu.ac.kr; Web: www.camc.re.kr Fax: +82-42-823-6665
First published on 27th October 2011
Abstract
Efficient and continuous monoacylation of symmetrical diamines performed in microreactors yielded superior selectivity to that predicted by statistical considerations. It is highly valuable that the kinetically controlled product in high yields was achieved without any special catalyst at ambient temperature.
Controlling product selectivity is an important and challenging research goal in most of the chemical reactions with multi-product environments. The product selectivity, often predicted by reaction kinetics, is achieved by controlling several parameters such as concentration, temperature, pressure, stoichiometry, nature and type of catalyst, auxiliary or solvents, etc. However in numerous chemical reactions, it has been observed that the amounts of side products are surprisingly very high despite of favourable reaction kinetics. In many cases, it has been demonstrated that the inefficient mixing in the batch reactors is a major barrier to achieve kinetically controlled products in such reactions.1
In competitive and consecutive chemical reactions, the product selectivity has been also an important issue to transform mono-functionalization. Selective monoacylation of symmetrical diamines is to provide monoacylated diamines that are key intermediates for the synthesis of numerous biologically active synthetic and natural products.2 Monoacylation of unsymmetrical diamines is relatively easier due to steric and/or electronic effects,3 whereas the same for symmetrical diamines has been found extremely challenging. Therefore, much attention has been focused on developing selective monoacylation of symmetrical diamines.4 Although many of these methods provide somewhat better selectivity, they suffer from their own limitations such as difficulties in using acylating agents having acidic protons in basic medium, use of aggressive and expensive reagents, difficulties in separation/purification of products, long reaction times, preparation of special acylating agents, exothermic heat dissipation problems, etc. Therefore, development of selective monoacylation of symmetrical diamines without any catalyst or special reagent will be highly valuable from a synthetic perspective.
Microreactors have recently attracted much interest in the scientific community for various research and applications.5 There is plenty of literature describing the use of capillary microreactors for the synthesis of valuable chemicals.6 In particular, the enhanced mixing and heat transfer of microreactors allowed to obtain kinetically favourable products as demonstrated in Friedel–Crafts alkylation and iodination of aromatic compounds, which were found very difficult to achieve in flask reactions.7 Furthermore, because the mixing and heat transfer become even more troublesome in large scale reactions as in pilot reactors, microreactors could be practical tools in such reactions to control reaction kinetics. Also, microreactors could be quite promising for achieving selective monoacylation of symmetrical diamines due to their inherent fast heat transfer capability and mixing.
Therefore, we initiated a systematic study with a simple model reaction in microreactors and flask reactors in a comparative manner. Firstly, reaction of 1,3-diaminopropane with N-hydroxy succinimide ester of benzoic acid (PhCOSu) was performed in the continuous capillary microreactor, droplet microreactor, and conventional bulk reactor as represented in Fig. 1. A PFA capillary (id 500 μm, length 50 cm and volume 98 μL) equipped with a T-micromixer was used as the continuous capillary microreactor. An alternative microreactor, fabricated by the PDMS and scaffold technique,8 was used for the droplet generation of diamine and acylating agent in methanol (dispersed phase) and consequent reaction by a droplet merging process in fluorocarbon oil (continuous phase).9 For comparison, the same reaction was also conducted in a 50 ml round bottom flask at identical conditions. Table 1 represents the results of selective monoacylation in the droplet, capillary microreactor and batch system.
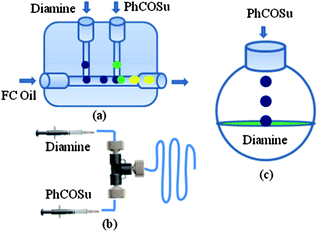 | ||
| Fig. 1 (a) Droplet microreactor, (b) continuous capillary microreactor and (c) batch reactor. | ||
|
|
||||
|---|---|---|---|---|
| Entry | Reactor | Res. time/min | Yield of 3aab (%) | Yield of 4aab (%) |
| a All the reactions were carried out at room temperature. b Isolated yield (1 mmol scale). c Standard drop-wise addition. d No drop-wise addition. e Ultrasonic agitation. | ||||
| 1 | Droplet | 5 | 55 | 39 |
| 2 | Droplet | 7 | 63 | 31 |
| 3 | Droplet | 10 | 61 | 32 |
| 4 | Capillary | 5 | 52 | 41 |
| 5 | Capillary | 10 | 55 | 39 |
| 6 | Batchc | 120 | 20 | 77 |
| 7 | Batchd | 10 | 5 | 92 |
| 7e | Capillary | 2 | 58 | 33 |
| 8e | Capillary | 1 | 61 | 33 |
| 9e | Capillary | 0.5 | 62 | 31 |
| 10e | Capillary | 0.33 | 52 | 29 |
| 11 | Capillary | 0.5 | 15 | 8 |
| 12e | Batchc | 120 | 19 | 74 |
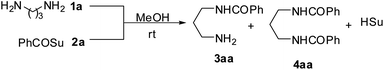
In the reaction of equimolar amounts of symmetrical diamine with the acylating agent, a mixture of monoacyl product, diacyl product and unreacted diamines would be formed in the molar ratio of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25:25 upon assuming the same reactivity of the amino groups in both the diamine and monoacyl product. And it has been reported that the rate of first acylation (monoacylation) is 3 to 15 times faster than second acylation,10 hence, theoretically expecting greater than 50% yield of the monoacyl product. However, the experimental result in the batch reactor showed the diacyl product 4aa as a major product and the monoacyl product 3aa as a minor product in the yield of 77:20 when a standard slow drop-wise addition protocol was followed for 2 h. In the case, the monoacyl product once formed in flask reaction (drop-wise addition) remained in touch with the incoming acylating agent and caused further acylation. Alternatively, the attempt to reduce reaction time (10 min) in the flask reaction by adding all the PhCOSu to the diamine at once yielded even worst result (4aa:3aa = 92:5), presumably due to inhomogeneous thermal condition with exothermic hot spots.
:25:25 upon assuming the same reactivity of the amino groups in both the diamine and monoacyl product. And it has been reported that the rate of first acylation (monoacylation) is 3 to 15 times faster than second acylation,10 hence, theoretically expecting greater than 50% yield of the monoacyl product. However, the experimental result in the batch reactor showed the diacyl product 4aa as a major product and the monoacyl product 3aa as a minor product in the yield of 77:20 when a standard slow drop-wise addition protocol was followed for 2 h. In the case, the monoacyl product once formed in flask reaction (drop-wise addition) remained in touch with the incoming acylating agent and caused further acylation. Alternatively, the attempt to reduce reaction time (10 min) in the flask reaction by adding all the PhCOSu to the diamine at once yielded even worst result (4aa:3aa = 92:5), presumably due to inhomogeneous thermal condition with exothermic hot spots.
However, both droplet and capillary microreactors revealed significantly better monoacylation selectivity than that in the batch reactor, even reversal of selectivity with the monoacyl product 3aa as a major product under identical reaction conditions (acylating agent, temperature, concentration), as shown in Table 1. In the batch system, although the stoichiometry of diamine and acylating agent is 1:1 on an average basis, it varies a lot as the drop-wise addition proceeds. On the other hand, in the case of microreactors, the stoichiometry of diamine to acylating agent is always constant (1:1). Different real time stoichiometry of diamine and acylating agents in batch and microreactors is expected to yield different selectivity in products. The striking performance of a microreactor against a batch reactor can be explained in a simple way by considering high mixing efficiency and heat dissipation capacity of the former. In the microreactor, it is known that the mixing driven by molecular diffusion leads to a homogeneous phase within a short time, and the reactions are quenched after precisely controlled retention time to protect the formed monoacylated product from incoming acylating agents. Since the heat of acylation is quickly dissipated due to inherent fast heat dissipating efficiency of the microreactor, the process virtually occurs under homogeneously isothermal conditions with no hot spots in contrast to what occurs in a flask (5–20% monoacylation). Thus, better temperature control in the microreactor helps to increase the product selectivity.11
Exactly speaking, the product compositions in this consecutive reaction were found to depend largely on the reaction times as well as the manner of mixing (entries 1, 2, 3 and entries 4, 5 in Table 1). The droplet microreactor showed slightly better product selectivity than the continuous capillary microreactor, it confirmed that the improved mixing efficacy with extra convection effect inside droplets enhanced the reaction performance of microreactors.12 However, the droplet as a dispersion phase was formed only in the presence of a continuous oil phase, which is not convenient for conventional organic reactions.
Therefore, it is appropriate to enhance the mixing by an alternative approach in the continuous capillary microreactor. Ultrasonication in microreactors has provided several advantages.13 Thus, the external ultrasonic agitation was chosen to perform the reaction by immersing the PFA capillary equipped with a T-micromixer into an ultrasonic bath (Table 1).
The monoacylated product was obtained up to 62% yield only for 0.5 min retention time (flow rate of 200 μL min−1) under ultrasonic irradiation, which is comparable with the droplet microreactor with 61% yield for 10 min reaction. The highest mono/diacyl selectivity (Table 1, entry 9) is in contrast to 15% yield of microreaction with no sonic agitation (Table 1, entry 11) as well as 19% yield of ultrasonic batch reaction for 2 h (Table 1, entry 12). It was obvious that the ultrasonic irradiation highly enhanced the mixing efficacy and accelerated the reaction for achieving high monoacylation selectivity.
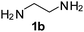
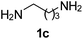
Monoacylation selectivity reversal was also accomplished through the reactions of various symmetrical diamines with RCOSu in an ultrasound promoted continuous flow capillary microreactor (Table 2). Mono- to diacylation selectivity was quite high in the case of piperazine (72:23), when compared with that of symmetrical primary diamines (around 2:1). In fact, piperazine and homopiperazine are very special as they are believed to stay as monoacetate salt in glacial acetic acid.14 Thus, they can be selectively monoacylated in glacial acetic acid to allow high selectivity. However, in batch reactors under conventional heating, the reactions were often incomplete even after prolonged reaction time and the yields of monoacylated product were only moderate. We, therefore, thought to investigate the monoacylation of piperazine in ultrasonic agitated capillary microreactors (Table 3).
| Diamine | R | Products | Selectivitya (monoacyl 3:diacyl 4) | |
|---|---|---|---|---|
| Microreactorb | Flaskc | |||
| a Based on isolated yields of monoacyl and diacyl products. b Residence time = 0.5 min; ultrasonication. c Ultrasonication 2 h. | ||||
|
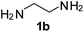
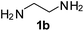
Ph, 2a | 3ba:4ba | 59:35 |
25:70 |
|
Ph, 2a | 3ca:4ca | 60:33 |
20:65 |
|
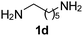
Ph, 2a | 3da:4da | 60:28 |
25:63 |
|
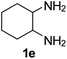
Ph, 2a | 3ea:4ea | 55:30 |
24:61 |
|
PhCH2, 2b | 3bb:4bb | 55:29 |
23:68 |
|
PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, 2c CH, 2c |
3bc : 4bc | 52:32 |
25:63 |
|
Ph, 2a | 3fa : 4fa | 72:23 |
40:55 |
|
|
||||
|---|---|---|---|---|
| Entry | Time/min | Temp./°C | Yield of 3fda (%) | Yield of 4fda (%) |
| a Isolated yield (1 mmol scale). b Reactions with ultrasonic irradiation. c Reactions without ultrasonic irradiation. d Literature value from ref. 14. NA = Not isolated. | ||||
| 1b | 2 | 25 | 25 | NA |
| 2b | 10 | 25 | 33 | NA |
| 3b | 2 | 60 | 91 | NA |
| 4b | 4 | 60 | 92 | NA |
| 5b | 2 | 80 | 91 | NA |
| 6c | 10 | 60 | 35 | NA |
| 7c | 480 | 60 | 56d | — |
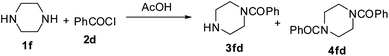
At room temperature (25 °C) the reaction was not complete and most of the piperazine was recovered as such even at low flow rates (Table 3, entries 1 and 2). By increasing the temperature, reaction became fast and 60 °C was found as an optimal temperature for the reaction. Further increasing the reaction temperature did not enhance the yield or decrease the reaction time to any significant extent (Table 3, entries 1–5). It is also obvious that reactions carried out under ultrasonic irradiation exhibited greater yields of monobenzoyl piperazine 3fd in comparison to those which were carried out without ultrasonic irradiation either in a microreactor or in a flask (Table 3, entries 6 and 7).
Ultrasonic irradiation appears to affect the yield and reaction time possibly by enhanced mixing of cavitation bubbles. In the glacial acetic acid, the need of heating to complete the reaction is not clear as the reaction of amine with the acylating agent is exothermic. In addition, there is no direct evidence for the existence of the piperazine completely as piperazine monoacetate in glacial acetic acid.14 However the results can be explained by assuming that piperazine and/or homopiperazine exists in equilibrium of diacetate and monoacetate salt in the glacial acetic acid. And upon heating the equilibrium is shifted towards monoacetate salt which in turn reacts with the acylating agent to complete the reaction.
In general, various aromatic, heteroaromatic as well as aliphatic acid chlorides gave excellent yields of monoacyl piperazine in the microreactor under ultrasonic irradiation (Table 4, entries 1–5). In none of the cases diacylpiperazine was obtained. Highly selective monoacylation of homopiperazine was also achieved with similar success and no diacyl homopiperazine was isolated in any experiment (Table 4, entries 6–8).
| Entry | Diamine | Acid chloride | Monoacyl product | Yieldb (%) |
|---|---|---|---|---|
| a Reaction with ultrasonic irradiation in a microreactor at 60 °C, residence time = 4 min. b Isolated yield (1 mmol scale). | ||||
| 1 | Piperazine 1f | PhCHCHCOCl 2e |
3fe | 88 |
| 2 | Piperazine 1f | 2-Thenoyl chloride 2f | 3ff | 90 |
| 3 | Piperazine 1f | 4-CH3C6H4SO2Cl 2g | 3fg | 93 |
| 4 | Piperazine 1f | CH3COCl 2h | 3fh | 90 |
| 5 | Piperazine 1f | PhCH2COCl 2i | 3fi | 87 |
| 6 | Homopiperazine 1g | PhCOCl 2d | 3gd | 89 |
| 7 | Homopiperazine 1g | PhCH2COCl 2i | 3gi | 91 |
| 8 | Homopiperazine 1g | 4-CH3C6H4SO2Cl 2g | 3gg | 94 |
Conclusions
In conclusion, we have demonstrated that symmetrical diamines can be selectively monoacylated in microreactors without using any additional catalysts or reagents. The use of ultrasonic irradiation in microreactors has synergistic effects over the yield as well as mono/diacyl selectivity. Piperazine and homopiperazine were found to be selectively monoacylated in glacial acetic acid under ultrasonic irradiation in excellent yields. The use of continuous capillary microreactors could be promising for mass production of monoacylated diamines because of its simplicity and high selectivity.Notes and references
- J.-i. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2011, 4, 331 CrossRef CAS.
- J. A. Bender, N. A. Meanwell and T. Wang, Tetrahedron, 2002, 58, 3111 CrossRef CAS.
- (a) T. Wang, Z. Zhang and N. A. Meanwell, Tetrahedron Lett., 1999, 40, 6745 CrossRef CAS; (b) T. Wang, Z. Zhang and N. A. Meanwell, J. Org. Chem., 2000, 65, 4740 CrossRef CAS; (c) T. R. Kelly and M. Cavero, Org. Lett., 2002, 4, 2653 CrossRef CAS.
- (a) Z. Zhang, Z. Yin, N. A. Meanwell, J. F. Kadow and T. Wang, Org. Lett., 2003, 5, 3399 CrossRef CAS; (b) T. Wang, Z. Zhang and N. A. Meanwell, J. Org. Chem., 1999, 64, 7661 CrossRef CAS; (c) W. Pringle, Tetrahedron Lett., 2008, 49, 5047 CrossRef CAS; (d) S. K. Verma, B. N. Acharya and M. P. Kaushik, Org. Lett., 2010, 12, 4232 CrossRef CAS; (e) B. P. Bandgar and S. S. Pandit, Tetrahedron Lett., 2003, 44, 3855 CrossRef CAS; (f) Y. Wang, J. Jin, M. L. Moore, T. L. Graybill, F. Wang, M. A. Wang, B. Wang, Q. Jin and R. A. Rivero, Tetrahedron Lett., 2004, 45, 6645 CrossRef CAS; (g) W. Tang and S. Fang, Tetrahedron Lett., 2008, 49, 6003 CrossRef CAS.
- (a) C. Wiles and P. Watts, Chem. Commun., 2011, 47, 6512 RSC; (b) J. Wegner, S. Ceylan and A. Kirschning, Chem. Commun., 2011, 47, 4583 RSC; (c) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502 CrossRef CAS; (d) R. A. Maurya, C. P. Park, J. H. Lee and D.-P. Kim, Angew. Chem., Int. Ed., 2011, 50, 5952 CrossRef CAS; (e) C. P. Park, R. A. Maurya, J. H. Lee and D.-P. Kim, Lab Chip, 2011, 11, 1941 RSC.
- (a) T. Noël, S. Kuhn, A. J. Musacchio, K. F. Jensen and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 5943 CrossRef; (b) P. B. Palde and T. F. Jamison, Angew. Chem., Int. Ed., 2011, 50, 3525 CrossRef CAS; (c) P. Salice, P. Maity, E. Rossi, T. Carofiglio, E. Menna and M. Maggini, Chem. Commun., 2011, 47, 9092 RSC; (d) P. P. Lange, L. J. Gooßen, P. Podmore, T. Underwood and N. Sciammetta, Chem. Commun., 2011, 47, 3628 RSC; (e) Y. Tomida, A. Nagaki and J.-i. Yoshida, J. Am. Chem. Soc., 2011, 133, 3744 CrossRef CAS.
- (a) K. Midorikawa, S. Suga and J.-i. Yoshida, Chem. Commun., 2006, 3794 RSC; (b) K. Kataoka, Y. Hagiwara, K. Midorikawa, S. Suga and J.-i. Yoshida, Org. Process Res. Dev., 2008, 12, 1130 CrossRef CAS; (c) A. Nagaki, M. Togai, S. Suga, N. Aoki, K. Mae and J.-i. Yoshida, J. Am. Chem. Soc., 2005, 127, 11666 CrossRef CAS; (d) S. Suga, A. Nagaki and J.-i. Yoshida, Chem. Commun., 2003, 354 RSC.
- A. Asthana, K.-O. Kim, J. Perumal, D.-M. Kim and D.-P. Kim, Lab Chip, 2009, 9, 1138 RSC.
- P. H. Hoang, C. T. Nguyen, J. Perumal and D. P. Kim, Lab Chip, 2011, 11, 329 RSC.
- A. R. Jacobson, A. N. Makris and L. M. Sayre, J. Org. Chem., 1987, 52, 2592 CrossRef CAS.
- T. Schwalbe, V. Autze, M. Hohmann and W. Stirner, Org. Process Res. Dev., 2004, 8, 440 CrossRef CAS.
- P. H. Hoang, H. Park and D.-P. Kim, J. Am. Chem. Soc., 2011, 133, 14765 CrossRef CAS.
- (a) T. Noël, J. R. Naber, R. L. Hartman, J. P. McMullen, K. F. Jensen and S. L. Buchwald, Chem. Sci., 2011, 2, 287 RSC; (b) J. Sedelmeier, S. V. Ley, I. R. Baxendale and M. Baumann, Org. Lett., 2010, 12, 3618 CrossRef CAS; (c) W. Shu, L. Pellegatti, M. A. Oberli and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, DOI:10.1002/anie.201105223; (d) S. Kuhn, T. Noël, L. Gu, P. L. Heider and K. F. Jensen, Lab Chip, 2011, 11, 2488 RSC.
- M. Desai, J. W. H. Watthey and M. Zuckerman, Org. Prep. Proced. Int., 1976, 8, 85 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and compound data. See DOI: 10.1039/c1lc20765b |
| This journal is © The Royal Society of Chemistry 2012 |