Structure and magnetic properties of ionic compound (Cp*2Cr+)·(FeIPc−)·(C6H4Cl2)4 containing negatively charged iron phthalocyanine†
Dmitri V.
Konarev
*a,
Leokadia V.
Zorina
b,
Salavat S.
Khasanov
b,
El'za U.
Hakimova
a and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow Region, 142432, Russia. E-mail: konarev@icp.ac.ru; Fax: +7-49652-21852
bInstitute of Solid State Physics RAS, Chernogolovka, Moscow Region, 142432, Russia
First published on 17th November 2011
Abstract
A new strategy for the preparation of molecular magnets by the reduction of phthalocyanines with strong metallocene donors has been suggested. Compound (Cp*2Cr+)·(FeIPc−)·(C6H4Cl2)4 (1) contains uniform stacks of alternating closely packed Cp*2Cr+ and FeIPc− ions. 1 is a soft ferrimagnet showing hysteresis with a small coercive field and a bifurcation temperature of about 5 K.
Phthalocyanines are widely used in the design of optical, magnetic and conducting materials.1 Conducting compounds were obtained by oxidation of phthalocyanines with iodine1b or electrochemical oxidation of the MIIIPc(CN)2− anions in the presence of organic cations.1c Magnetic compounds were obtained by oxidation of manganese(II) phthalocyanine (MnIIPc) or octa-n-butoxynaphthalocyanine (MnIINc(OBu)8) by tetracyanoethylene (TCNE). The TCNE˙−radical anions directly coordinate to the MnIII atoms of phthalocyanine by cyano groups to form coordination chains of alternating MnIIIPc+ or MnIIINc(OBu)8+ and TCNE˙− ions.1d,e MnPc·TCNE shows ferromagnetic coupling without magnetic ordering,1d whereas MnNc(OBu)8·TCNE is a ferrimagnet in which MnIIINc(OBu)8+ (S = 2) and TCNE˙− (S = 1/2) ions antiferromagnetically interact in the chains.1e All conducting and magnetic compounds of phthalocyanines known now have been obtained by oxidation.
Phthalocyanines can also be reduced to form anions with charged state from −1 to −4.2 Among different phthalocyanines FeIIPc has slightly lower reduction potential (−0.70 V vs.SCE in non-coordinating 1-methylnaphthalene).3 It is an essentially weaker acceptor than TCNE (E0/− = +0.29 V),4bis-dichalcogenate acceptors, for example, Ni(dmit)2 (dmit: 1,3-dithiole-2-thione-4,5-dithiolate) (E0/− = −0.08 V),5 and even fullerene C60 (E0/− = −0.44 V).4Decamethylchromocene (Cp*2Cr, E+/0 = −1.04 V vs.SCE)6 forms ionic complexes with all these acceptors and some of the complexes with Cp*2Cr+ exhibit interesting magnetic properties: (Cp*2Cr+)·(TCNE˙−) is a ferromagnet with Tc of 3.65 K;7 the complexes with some bis-dichalcogenate acceptors manifest metamagnetic behavior;8 a magnetic transition is observed in (Cp*2Cr+)·(C60−)·(C6H4Cl2)2 due to the reversible formation of the diamagnetic singly bonded (C60−)2 dimers.9 According to redox potential Cp*2Cr is also a strong enough donor to reduce FeIIPc and form an ionic compound with this phthalocyanine. At present only salts of FeIPc− with potassium cryptand or lithium crown ether have been obtained and structurally characterized. For their preparation FeIIPc was directly reduced by metals in the presence of cryptand or crown ether in tetrahydrofuran.10
Here we show for the first time that strong metallocene donors like decamethylchromocene can also be used for preparation of ionic compounds with metal phthalocyanines. One compound (Cp*2Cr+)·(FeIPc−)·(C6H4Cl2)4 (1) showing interesting magnetic properties is obtained and its synthesis, crystal structure and magnetic properties are discussed.
Neutral FeIIPc only weakly dissolved in the C6H4Cl2/C6H5CN mixture (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). The addition of 1.4 molar equivalents of Cp*2Cr under anaerobic conditions resulted in complete dissolution of FeIIPc due to reduction and a deeply colored red-violet solution was formed. The use of a slight excess of Cp*2Cr is possible since FeIIPc has very negative second reduction potential (−1.13 V vs.SCE)3 and cannot be reduced by Cp*2Cr to the dianionic state. Well-shaped elongated parallelepipeds were obtained by the diffusion of hexane into this solution during 6 weeks. The solvent was decanted and the crystals were washed with hexane (the yield was about 10%). Composition of 1 was determined from X-ray diffraction on a single crystal. Several crystals tested from the synthesis had the same unit cell parameters which showed that they belonged to one crystal phase.
:3). The addition of 1.4 molar equivalents of Cp*2Cr under anaerobic conditions resulted in complete dissolution of FeIIPc due to reduction and a deeply colored red-violet solution was formed. The use of a slight excess of Cp*2Cr is possible since FeIIPc has very negative second reduction potential (−1.13 V vs.SCE)3 and cannot be reduced by Cp*2Cr to the dianionic state. Well-shaped elongated parallelepipeds were obtained by the diffusion of hexane into this solution during 6 weeks. The solvent was decanted and the crystals were washed with hexane (the yield was about 10%). Composition of 1 was determined from X-ray diffraction on a single crystal. Several crystals tested from the synthesis had the same unit cell parameters which showed that they belonged to one crystal phase.
Crystallographic data were collected at 150(1) K.11 The tested crystals were non-merohedral twins. Diffraction reflections from both twin domains were used in the structure refinement (SHELX HKLF5 file). The twin matrix was (1 0 0, −2/3 −1 0, −1 0 −1); the refined twin fraction in the studied crystal was 0.487(1). Phthalocyanine is ordered in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) lattice, whereas the Cp*2Cr+ cations are disordered between two orientations with the 0.6/0.4 occupancies. These orientations are linked by the rotation of the cation by ∼17° about a five-fold axis of Cp*2Cr. The complex contains two crystallographically independent positions of solvent C6H4Cl2 molecules in which they are disordered between three orientations. Solvent and Cp*2Cr+ are shown in Fig. 1 in major orientation. The main structural motif of crystal structure of 1 is uniform stacks of alternating Cp*2Cr+ and FeIPc− ions (Fig. 1a). Both iron and chromium metal atoms are positioned on the inversion centers and the Fe(I)–Cr(III) distances in the stacks are equal to a/2 = 5.27 Å. The stacks are packed in a plane-to-plane manner: Cp*2Cr+ ions are sandwiched between FeIPc− ions. Phthalocyanine has a flattened conformation. There is a π–π interaction between phthalocyanine and the Cp* planes of Cp*2Cr+. The interplanar distance is equal to 3.46 Å and these planes are located nearly parallel to each other (the corresponding dihedral angle is only 1.5°). There are short Fe(FeIPc−)⋯C(Cp*) contacts in the 3.49–3.67 Å range (Fig. 2) and multiple short contacts (2.65–2.76 Å) are formed between hydrogen atoms of methyl groups of the Cp* rings and C and N atoms of the phthalocyanine plane. These contacts were calculated for major orientation of Cp*2Cr+.
lattice, whereas the Cp*2Cr+ cations are disordered between two orientations with the 0.6/0.4 occupancies. These orientations are linked by the rotation of the cation by ∼17° about a five-fold axis of Cp*2Cr. The complex contains two crystallographically independent positions of solvent C6H4Cl2 molecules in which they are disordered between three orientations. Solvent and Cp*2Cr+ are shown in Fig. 1 in major orientation. The main structural motif of crystal structure of 1 is uniform stacks of alternating Cp*2Cr+ and FeIPc− ions (Fig. 1a). Both iron and chromium metal atoms are positioned on the inversion centers and the Fe(I)–Cr(III) distances in the stacks are equal to a/2 = 5.27 Å. The stacks are packed in a plane-to-plane manner: Cp*2Cr+ ions are sandwiched between FeIPc− ions. Phthalocyanine has a flattened conformation. There is a π–π interaction between phthalocyanine and the Cp* planes of Cp*2Cr+. The interplanar distance is equal to 3.46 Å and these planes are located nearly parallel to each other (the corresponding dihedral angle is only 1.5°). There are short Fe(FeIPc−)⋯C(Cp*) contacts in the 3.49–3.67 Å range (Fig. 2) and multiple short contacts (2.65–2.76 Å) are formed between hydrogen atoms of methyl groups of the Cp* rings and C and N atoms of the phthalocyanine plane. These contacts were calculated for major orientation of Cp*2Cr+.
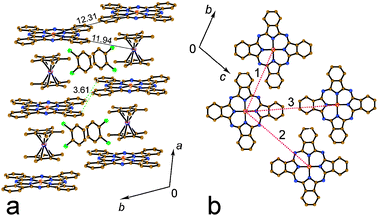 | ||
| Fig. 1 View of crystal structure of 1. Two stacks from the alternating Cp*2Cr+ and FeIPc− ions are viewed along the c-axis (a); view along the a-axis and the Cp*2Cr+–FeIPc− stacks (b). Solvent C6H4Cl2 molecules located between the stacks are also shown. Dashed lines indicate distances between iron atoms of FeIPc− belonging to neighboring stacks: (1) 12.31; (2) 14.97 and (3) 15.78 Å. | ||
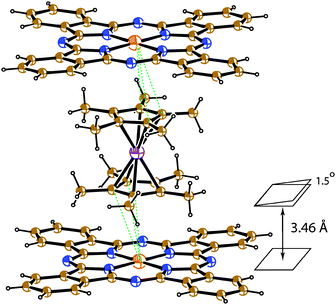 | ||
| Fig. 2 van der Waals contacts between Cp*2Cr+ and FeIPc− ions (dashed lines). | ||
There are large voids in the packing of the Cp*2Cr+–FeIPc− stacks occupied by the C6H4Cl2 molecules. Totally each Cp*2Cr+ is surrounded by four C6H4Cl2 molecules located between the phenylene groups of two phthalocyanines. As a result, the Cp*2Cr+–FeIPc− stacks are isolated by solvent molecules and there are no direct vdW contacts between them. The shortest C(FeIPc−)⋯C(FeIPc−) contact of 3.61 Å is formed along the b-axis (Fig. 1a). Each stack is surrounded by six neighboring stacks. The shortest Fe⋯Fe distances for phthalocyanines from neighboring stacks are large enough varying from 12.31 to 15.78 Å (these distances are shown by red dashed lines in Fig. 1b). Therefore, short Fe(I) and Cr(III) distances are observed within the stacks, whereas the interstack distances between metal atoms are essentially longer.
The charge state of Cp*2Cr in 1 can be estimated from the IR-spectrum (ESI†). The bands at 436 w, 548 w, 1025 w and 1380 m cm−1 are ascribed to Cp*2Cr. The shift of the band of neutral Cp*2Cr at 418 cm−1 to 436 cm−1 in the spectrum of 1 indicates the formation of Cp*2Cr+. A similar shift of this band to 437 cm−1 is observed in the spectra of ionic (Cp*2Cr+)·(PF6−)6 and (Cp*2Cr+)·(C60−)·(C6H4Cl2)2.9
Magnetic properties of 1 were studied by SQUID in the 2–300 K range for two samples from different syntheses. Both samples show similar magnetic behavior. The contributions from the holder and core temperature independent susceptibility were subtracted from the experimental data. Above 80 K reciprocal magnetic susceptibility can be fitted well by the Curie–Weiss expression with a negative Weiss temperature of −10 K (Fig. 3, inset) indicating that antiferromagnetic interaction (AF) of spins dominates at high temperatures. Below 80 K reciprocal magnetic susceptibility deviates from the Curie–Weiss law to the ferromagnetic side (Fig. 3, inset). Effective magnetic moment of the complex is equal to 4.20 μB at 300 K. This value is close to that calculated for the system of two non-interacting S = 3/2 (Cp*2Cr+)6 and S = 1/2 spins per formula unit (μeff = 4.24 μB). Previously it was shown that reduction of FeIIPc is metal-centered and is accompanied by the formation of FeIPc− with S = 1/2 spin state.2,12 Magnetic moment slightly decreases with temperature attaining the minimum at Tm = 80 K (μeff = 4.08 μB) and magnetic moment increases rapidly below this temperature (Fig. 3) indicating that ferromagnetic interaction of spins (FM) dominates below 80 K. The minimum on the temperature dependence of the effective magnetic moment is characteristic of 1D antiferromagnetic coupling.13
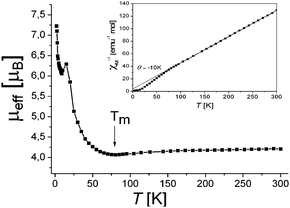 | ||
| Fig. 3 Temperature dependence of effective magnetic moment and reciprocal magnetic susceptibility (inset) for 1 in the 2–300 K range. | ||
Magnetization is nearly saturated at high fields and the value of 14766 emu Oe mol−1 is observed at 5T (ESI†). This value is substantially lower than the expected saturation magnetization value for a ferromagnetically coupled system of 22340 emu Oe mol−1 (Stot = 3/2 + 1/2 = 2) and slightly higher than the expected saturation magnetization value for an antiferromagnetically coupled system of 11170 emu Oe mol−1 (Stot = 3/2 − 1/2 = 1). The observed value is closer to the antiferromagnetically coupled Stot = 3/2 − 1/2 = 1 system. Therefore, 1 can be a ferrimagnet. Higher saturation magnetization values that can be calculated were previously observed in some 1-D ferrimagnetic chain compound, for example, MnNc(OBu)8·TCNE.1e
Fig. 4a shows field-cooled magnetization (FCM), zero-field-cooled magnetization (ZFCM), and remanent magnetization (inset in Fig. 4a) for 1 measured in the 2–30 K temperature range. When the field is switched off at 2 K, remanent magnetization is observed. It nearly vanishes as temperature increases to 5 K. The ZFCM also merges with the FCM curve as temperature is raised above 5 K. Thus bifurcation temperature for 1 is about 5 K.
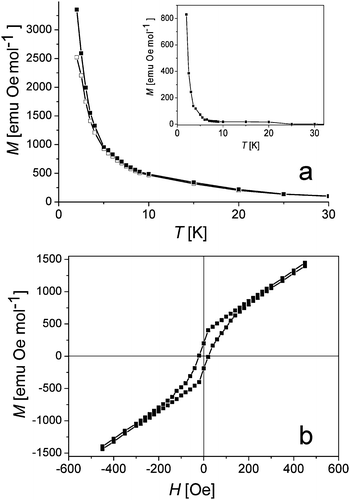 | ||
| Fig. 4 (a) Plot of zero-field-cooled magnetization (ZFCM) (open squares), field-cooled magnetization (FCM) (black squares) in 1000 Oe, remanent magnetization vs. temperature for 1 (inset); (b) the M(H) hysteresis loop at 2 K. | ||
Compound 1 can be classified as a soft ferrimagnet since it shows hysteresis with small coercive field, Hcr, of 20 Oe and small remanent magnetization of 200 emu Oe mol−1 (Fig. 4b). The absence of hysteresis or small coercive field is characteristic of magnets based on the Cp*2Cr+ cations. For example, hysteresis was neither found in ferromagnet (Cp*2Cr+)·(TCNE˙−). That was ascribed to the lack of anisotropy at the Cr(III) center.8
In this work we for the first time used strong metallocene donor decamethylchromocene to prepare an ionic compound with negatively charged iron phthalocyanine (Cp*2Cr+)·(FeIPc−)·(C6H4Cl2)4 (1). The structure of 1 involves uniform stacks of closely packed alternating Cp*2Cr+ and FeIPc− ions nearly isolated by solvent C6H4Cl2 molecules. Magnetic measurements show that AF interaction dominates in 1 above 80 K, whereas FM interaction is observed at T < 80 K. The compound is a soft ferrimagnet showing hysteresis with a small coercive field. It has a bifurcation temperature of about 5 K. Metallocenes form a variety of magnetic compounds with organic or bis-dichalcogenate acceptors.7,8 The presented strategy for the reduction of phthalocyanines by strong metallocene donors allows one to extend the family of molecular-based magnets by using metal-containing and metal-free phthalocyanines. The anionic complex of metal-free phthalocyanine H2Pc with Cp*2Cr has also been synthesized by us and its crystal structure and magnetic properties will be presented elsewhere.
Experimental
Materials
Cp*2Cr, FeIIPc was purchased in Aldrich. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 in an argon atmosphere under vacuum. Benzonitrile was distilled over Na/benzophenone under vacuum. Hexane was doubly distilled over Na/benzophenone in an argon atmosphere. The solvents were degassed and were put into a glove box.General
All manipulations on the synthesis and the isolation of the crystals were carried out in the glove box MBraun 150B-G with a controlled atmosphere and the content of H2O and O2 less than 1 ppm. FT-IR spectra were measured in KBr pellets with a Perkin-Elmer 1000 Series spectrometer (400–7800 cm−1). The preparation of KBr pellets for IR-measurements was carried out in a glove box. Magnetic susceptibility measurements were carried out with a Quantum Design MPMS-XL SQUID magnetometer at a magnetic field of 1000 G between 2 and 300 K under zero-field and field (1000 G) cooling conditions.Syntheses
Iron(II) phthalocyanine (FeIIPc, 25 mg, 0.044 mmol) is added to 15 mL of the C6H4Cl2/C6H5CN mixture (12:3). It dissolved very weakly in this mixture forming slightly colored green solution. The addition of Cp*2Cr (20 mg, 0.062 mmol) results in complete dissolution of FeIIPc within minutes due to the reduction with the formation of deeply colored red-violet solution. Solution is stirred for 2 hours and is filtered into a glass tube of 1.5 cm diameter and 40 mL volume with a ground glass plug. Hexane (20 mL) is carefully layered. The crystals of 1 were prepared by diffusion of hexane during 6 weeks. They form on the walls of the tube as thin elongated parallelepipeds. The solvent was decanted and the crystals were washed with hexane yielding crystals up to 2 × 0.2 × 0.1 mm3 size with only 10% yield. The composition of (Cp*2Cr)·(FePc)·(C6H4Cl2)4 (1) was determined by the X-ray diffraction on a single crystal. Several crystals from the synthesis were checked by the X-ray diffraction and have the same unit cell parameters as 1.
Acknowledgements
This work was supported by the RFBR Grant No. 09-02-01514.Notes and references
- (a) T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707 CrossRef CAS; (b) T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS; (c) H. Hasegawa, T. Naito, T. Inabe, T. Acutagawa and T. Nakamura, J. Mater. Chem., 1998, 8, 1567 RSC; (d) J. S. Miller, C. Vazquez, J. C. Calabrese, M. L. McLean and A. J. Epstein, Adv. Mater., 1994, 6, 217 CrossRef CAS; (e) D. K. Rittenberg, L. Baars-Hibbe, A. B. Böhm and J. S. Miller, J. Mater. Chem., 2000, 10, 241 RSC.
- A. B. P. Lever, E. P. Milaeva and G. Speier, In Phthalocyanines, properties and applications, ed. C. C. Lezuoff and A. B. P. Lever, VCH, New York, 1993, vol. 3, pp. 1–70 Search PubMed.
- R. H. Campbell, G. A. Heath, G. T. Hefter and R. C. S. McQueen, J. Chem. Soc., Chem. Commun., 1983, 1123 RSC.
- G. Saito, T. Teramoto, A. Otsuka, Y. Sugita, T. Ban, M. Kusunoki and K.-I. Sakaguchi, Synth. Met., 1994, 64, 359 CrossRef CAS.
- R. Kato, Chem. Rev., 2004, 104, 5319 CrossRef CAS.
- J. L. Robbins, N. Edelstein, B. Spencer and J. C. Smart, J. Am. Chem. Soc., 1982, 104, 1882 CrossRef CAS.
- J. S. Miller, R. S. Mclean, C. Vazquez, J. C. Calabrese, F. Zuo and A. J. Epstein, J. Mater. Chem., 1993, 3, 215 RSC.
- V. Gama and M. T. Duarte, In Magnetism: Molecules to materials V, ed. J. S. Miller and M. Drillon, Wiley-VCH, Weinheim, 2005, pp. 1–40 Search PubMed.
- (a) D. V. Konarev, S. S. Khasanov, A. Otsuka and G. Saito, J. Am. Chem. Soc., 2002, 124, 8520 CrossRef CAS; (b) D. V. Konarev, S. S. Khasanov, G. Saito, A. Otsuka, Y. Yoshida and R. N. Lyubovskaya, J. Am. Chem. Soc., 2003, 125, 10074 CrossRef CAS.
- M. Tahiri, P. Doppelt, J. Fischer and R. Weiss, Inorg. Chim. Acta, 1987, 127, L1 Search PubMed.
-
Crystal data of 1: C76H62Cl8CrFeN8, Mr = 1478.79, triclinic, P, a = 10.5331(12), b = 12.3100(11), c = 14.9616(13) Å, α = 98.283(7)°, β = 110.587(9)°, γ = 106.472(9)°, V = 1676.1(3) Å3, Z = 1, ρcalcd = 1.465 g cm−3, T = 150(1) K, data/parameters/restrains 17996/522/455, R1 = 0.0974, wR2 = 0.2338, G.O.F. = 1.071. CCDC 842064.
- T. Kaczmarzyk, T. K. Jackowski and K. Dziliński, Nukleonika, 2007, 52, S99 Search PubMed.
- E. Coronado, M. Drillon and R. Georges, In Research Frontiers in Magnetochemistry, ed. C. O'Connor, World Scientific Publishing, Singapore, 1993, pp. 27–66 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: details of crystal structure refinement and IR-spectrum of 1. CCDC 842064. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c1nj20858f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |