The adsorption of an anticancer hydrazone by protein: an unusual static quenching mechanism†
Fang-Fang
Tian
,
Jia-Han
Li
,
Feng-Lei
Jiang
,
Xiao-Le
Han
,
Chen
Xiang
,
Yu-Shu
Ge
,
Li-Li
Li
and
Yi
Liu
*
State Key Laboratory of Virology & Key Laboratory of Analytical Chemistry for Biology and Medicine (MOE), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, P. R. China. E-mail: yiliuchem@whu.edu.cn; Fax: +86 27 68756667
First published on 16th November 2011
Abstract
A novel hydrazone, 4-chloro-N′-(pyridin-2-ylmethylene)benzohydrazide (CPBH) has been synthesized through a one-pot synthesis method and used as a chemical probe to find the structural cause of the unusual static quenching mechanism in the interaction with serum albumin. The adsorption of CPBH by bovine/human serum albumin (BSA/HSA) has been investigated systematically by comprehensive spectroscopy, modeling, electrochemistry and microcalorimetry under physiological conditions. CPBH forms a complex with BSA/HSA with the binding site in Sudlow's site I of BSA/HSA. The adverse temperature dependence in the unusual static quenching is found to be a reasonable consequence of the large activation energy requirement in the binding process, which is required to overcome the structural block and it is a direct result of the unique microstructure of the binding pocket.
1. Introduction
The adsorption of drugs by serum albumin (SA) is crucial for the drugs' transportation and distribution.1–4 Serum albumin is the most abundant protein in blood plasma and responsible for the maintenance of osmotic pressure and transportation of various endogenous and exogenous compounds, including nutriment, pharmaceuticals and harmful substances.5–7 Serum albumin has been studied extensively due to its importance in biology, stability and availability. The most widely used albumins are bovine and human serum albumin.1–7As previously studied, human serum albumin (HSA) is a 585 amino acid residue monomer made up of three homologous α-helical domains.8 There are two common binding sites called Sudlow I and Sudlow II in HSA, which are hydrophobic cavities located in sub-domains IIA and IIIA.9 Moreover, the tryptophan residue we are concerned about in fluorescence spectroscopy is located in Sudlow I (Trp214).4,8 The hydrophobic cavities are critical binding sites for hydrophobic drugs in plasma as they increase the apparent solubility of hydrophobic drugs.10,11 Bovine serum albumin (BSA) is homologous with HSA, sharing a homology similarity of 88% in their amino acid sequences, so they have almost the same shape and binding sites.4,10 Studies on serum albumin and recently discovered and synthesized drugs are imperative to provide information about the absorption and uptake of these drugs. Because of the good fluorescence performance of tryptophan in SA, fluorescence spectroscopy is employed broadly in this study about albumin.
Fluorescence quenching spectroscopy is a special form of fluorescence spectroscopy and mainly based on the quenching behavior of drugs towards fluorescent substances.12,13 If the interaction between the drug and albumin can induce the quenching of the intrinsic fluorescence emission of albumin, we can conduct a fluorescence quenching titration experiment to get information about the binding properties of the drug–albumin system, through the quenching mechanism and the detailed quenching behavior.
As described, the quenching mechanism of a protein's fluorescence emission can be classified as static quenching and dynamic quenching, and they represent two totally different quenching processes.14–17 The former is caused by the ground state complex formation between drug and protein, while the latter is a result of the collision between the excited fluorescent molecules and the drug molecules.4,10,18,19 In both static and dynamic quenching, the percentage of the excited fluorescent molecules reduces, thus leading to the quenching of emission.17,20
These two kinds of quenching mechanism demonstrate some differences that can be distinguished experimentally, such as the change in the UV-visible spectra of the protein and the temperature dependence of the quenching constant.17,20,21 A complex of protein and drug forms in static quenching, so there will be some changes in the UV-visible spectra of the protein, whereas dynamic quenching has no such change. As to the temperature dependence of the quenching constant, diffusion is the control step for dynamic quenching, so the quenching constant will increase with increasing temperature.22 Furthermore, the adverse effect of increasing temperature can be detected in static quenching, as the stability of the complex will decrease with increasing temperature.17,20,22 As reported by our group and other research groups, most of the quenching is in good accordance with this theory and fits the results in the distinguishing experiments.4,5,23–25 However, as some exceptions appeared when studying the interactions between some novel acylhydrazone derivatives and serum albumin in our group,10 and in other reports,46,47 a doubt has been raised. Can this theory cover all the quenching behaviors of drugs towards protein, or in another words, do all the quenching behaviors exactly fit the principle described in this theory? What is the determinant factor if the answer is no? Does the particularity of the hydrazones' structure cause the exception? Or maybe something unknown causes a deviation from the principle above.
Hydrazones are a class of organic compounds with the common structure, R1R2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NNR3R4, and are usually prepared via the condensation of aldehydes or ketones with hydrazines in the presence of an acid catalyst.26Hydrazones have received much attention because of their particular physical, chemical and biological activities based on their unique structure.26 As reported, hydrazones have been widely employed as leading compounds in pharmacy,27,28catalysts for organic synthesis,29,30 basic function groups in optoelectronic materials,31 linkers in the interface sciences,32 and so on. Recently, chemists have shown much interest in the potential application of hydrazones in pharmacy as anti-bacterial33, anti-tumor34–37, anti-HIV38, anti-convulsant39 and anti-tubercular40 agents.
NNR3R4, and are usually prepared via the condensation of aldehydes or ketones with hydrazines in the presence of an acid catalyst.26Hydrazones have received much attention because of their particular physical, chemical and biological activities based on their unique structure.26 As reported, hydrazones have been widely employed as leading compounds in pharmacy,27,28catalysts for organic synthesis,29,30 basic function groups in optoelectronic materials,31 linkers in the interface sciences,32 and so on. Recently, chemists have shown much interest in the potential application of hydrazones in pharmacy as anti-bacterial33, anti-tumor34–37, anti-HIV38, anti-convulsant39 and anti-tubercular40 agents.
Hydrazones possess very good bioactivities, but how they are absorbed and transported to a specific location still remains unknown. However, as we mentioned above, the absorption, distribution and metabolism of drugs are strongly dependent upon the binding properties towards SA.1–4 So the systematic investigation of the interactions between SA and hydrazone is very important and obligatory to understand the detailed pharmacology and pharmacokinetics for drugs.
In this paper, we have designed and synthesized a novel hydrazone, 4-chloro-N′-(pyridin-2-ylmethylene)benzohydrazide (CPBH), an analogue of 311 ((E)-N′-((2-hydroxynaphthalen-1-yl)methylene)benzohydrazide) and PIH ((E)-N′-((3-hydroxy-5-(hydroxymethyl)-2-methylpyridin-4-yl)methylene)isonicotinohydrazide) which have both been used as potential anticancer drugs.41 We have comprehensively studied its interactions with two model proteins, BSA and HSA, by spectroscopy, electrochemistry, molecular docking and microcalorimetry. We focus on the relationship between structure factors and the binding mechanism of albumin towards CPBH, which has a great impact on the binding properties.
2. Experimental section
2.1 Reagent and apparatus
The reactants and solvent used in the synthesis and analysis were purchased from Sinopharm Chemical Reagent Co. Ltd of AR grade and used without further purification. BSA and HSA, purchased from Sigma Aldrich, were prepared in PBS (pH = 7.4), and then kept in a refrigerator at 4 °C. CPBH was dissolved in ethanol at the concentration of 20 mM as stock solution, then diluted with PBS before use. Water used in all the procedures was obtained from a Millipore water purification system.A LS-55 spectrofluorophotometer, equipped with a thermostat bath from Perkin Elmer, was employed to record the fluorescence spectra of the SA and CPBH system. A UNICO 4802 UV-vis Double Beam Spectrophotometer was used to measure absorption spectra. The electrochemical experiments were performed on a CHI660C electrochemical workstation from Shanghai Chenhua Instrument Company. A nano-ITC2G from TA Instruments was used to study the binding interaction process. For the synthesis and identification, elemental analysis was performed with an Elementar Aralysensysteme GmbH VarioEL elemental analysis system. Infrared spectra were measured on an Avatar 360 FT-IR spectrophotometer as KBr pellets.
2.2 Preparation
As shown in Scheme 1, a one-pot synthesis method had been developed to obtain the target substance CPBH. Hydrazine hydrate (85%, 0.11 mL, 2 mmol, in 5 mL ethanol) was added to a solution of methyl-4-chlorobenzoate (0.34 mL, 2 mmol) in ethanol (20 mL), using acetic acid as a catalyst. After the mixture was heated and stirred at 90 °C for 4 h, a solution of picolinaldehyde (0.214 g, 2 mmol, dissolved in 10 mL of ethanol) was added to this stirred solution. The mixture was refluxed for another 4 h, then cooled down to room temperature overnight. The precipitant was collected and washed, then recrystallized from ethanol. A white powder was obtained with a total yield of 93%. The elemental analysis results are: Found (Calc.): C 60.61 (60.12); H 3.62 (3.88); N 16.32 (16.18). 1H NMR (300 M, in DMSO-d6): δ = 8.57 ppm (d, 1H, 6-H in pyridyl), 8.39 (s, 1H, NCH), 8.31 (d, 1H, 3-H in pyridyl), 7.96 (d, 2H, 2,6-H in Ar), 7.90 (m, 1H, 4-H in pyridyl), 7.55 (d, 2H, 3,5-H in Ar), 7.44 (t, 1H, 5-H in pyridyl). IR: 3410 cm−1 (m, O-H), 3250 (m, C–H), 1670 (s, CO), 1620 (m, imide II), 1600 (s, CN), 1540, 1470, 1430 (m, Ar), 1270 (s, CN). UV-visible (in PBS; L mol−1 cm−1): 196.0 nm (4.90 × 104); 240.0 nm (2.28 × 104); 299.0 nm (5.20 × 104).
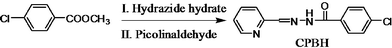 | ||
| Scheme 1 One-pot synthesis of CPBH. | ||
2.3 Spectra measurement
The fluorescence spectra were measured with a 1 cm quartz cell at 313, 308, 303 and 298 K, using 15 nm/6 nm slit widths in all experiments. The excitation and emission wavelengths for SA were 280 nm and 351 nm, respectively. Very dilute solutions of SA (2 μM) and CPBH (0–5 μM) were used to avoid inner filter effects.4,10,18 The site-competitive replacement experiments were conducted by adding CPBH to a SA-site marker system.4,10,18,23,44 The UV-visible absorption spectra were measured with a 1 cm quartz cell.2.4 Electrochemical experiments
The electrochemical experiments were conducted using a three-electrode electrochemical system. The working electrode was a gold disk electrode (diameter = 2 mm), while a Ag/AgCl electrode served as a reference electrode with a Pt wire counter electrode. The preparation of a SA-modified working electrode was based on the dry adsorption method developed in former work.10,42The electrolyte contained 5 mM K3Fe(CN)6/K4Fe(CN)6 and 10 mM KCl at pH 7.4. 10 mL electrolyte was used in each test. Various volumes of CPBH solutions were added continuously to the system and stirred for 2 min and then rested for 2 min before testing. The electrochemical impedance spectroscopy (EIS) were measured with the frequency from 0.1 to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz, and then analyzed by a Z-view software to obtain the Rct.42
000 Hz, and then analyzed by a Z-view software to obtain the Rct.42
2.5 Molecular docking investigation
The structure of CPBH was drawn using the Sybyl 8.1 package. The molecule was charged using the Gasteiger–Marsili method, without changing the formal charge before computation. Then the molecule of CPBH was optimized in energy and geometry using the Powell method. The detailed settings were as follows: Initial Optimization: Simplex; Termination: Gradient at 0.01 kcal mol−1; Force Field: Tripos; Other parameters were using the default setting.The 3D structure of BSA used in the docking studies was modeled according to the literature,10 while the PDB entry of the HSA crystal structure employed in docking studies was 1H9Z. The HSA crystal structure contained a warfarin and some fatty acid molecules which were extracted before docking. Docking studies were conducted usng a Surflex Dock program in the Sybyl 8.1 package. The protomol was generated by the Residues mode for BSA and Ligand mode for HSA with warfarin as the ligand. The Threshold was set at 0.50, while the Bloat was 0. The parameters in docking work was set as follows: Additional Starting Conformation per Molecule: 20; Angstroms to Expand Search Grid: 6; Max Conformation per Fragment: 20; Max Number of Rotatable Bonds per molecule: 100.
2.6 Microcalorimetry experiments
The isothermal titration was performed as an Incremental Titration with a 1000 μL cell and a 250 μL syringe. All the experiments was conducted at 25 °C, while the stir speed was set to 200 rpm. The time settings in the titration were as following: Data Interval: 1.0 s; Injection Interval: 300 s for HSA and 600 s for BSA; Start Delay: 1800 s; Initial Baseline: 900 s for HSA and 1800 s for BSA. The injection number was 20 and the injection volume for 2–20th injection was 12 μL whereas the first injection volume was set as 5 μL. The control experiment for each system was conducted under the same conditions, using CPBH as titrant and PBS as the titrand.The baseline of the thermogram was manually set and then the peak area was integrated using NanoAnalyze software provided by TA Instruments. Subtracting the control experiment in Average Area mode gave the corrected reaction heat for each injection. Then we used the Independent mode to fit the data to get the parameters for the binding process.
3. Results and discussion
3.1 Quenching mechanism of CPBH-SA
Fluorescence spectroscopy is widely used in biochemical, medical, and chemical research fields.14,43 We can get different information according to different data such as emission spectra, excitation spectra, quantum yield, polarization and fluorescent lifetime.14,43 So we applied fluorescence spectroscopy in studying the interactions between CPBH and SA.Fluorescence titration measurements were carried out to examine whether CPBH was adsorbed to SA. The emission spectra of SA in the absence and presence of CPBH are presented in Fig. 1. It can be clearly seen that both BSA and HSA show a strong emission band at 351 nm when the excitation wavelength is fixed at 280 nm, while CPBH shows almost no emission under the same conditions. The fluorescence intensity of SA decreases regularly and acutely with an increasing concentration of CPBH, which means that CPBH quenches the intrinsic fluorescence of SA and this effect positively correlates to the concentration of CPBH.
![Effect of CPBH on the fluorescence spectra of [A] BSA and [B] HSA at 298 K. c(BSA/HSA) = 2 μM; c(CPBH)/μM, A–P: 0; 0.33; 0.67; 1.00; 1.33; 1.67; 2.00; 2.33; 2.67; 3.00; 3.33; 3.67; 4.00; 4.33; 4.67; 5.00. The curve at the bottom shows the emission spectrum of CPBH only under this condition.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f1.gif) | ||
| Fig. 1 Effect of CPBH on the fluorescence spectra of [A] BSA and [B] HSA at 298 K. c(BSA/HSA) = 2 μM; c(CPBH)/μM, A–P: 0; 0.33; 0.67; 1.00; 1.33; 1.67; 2.00; 2.33; 2.67; 3.00; 3.33; 3.67; 4.00; 4.33; 4.67; 5.00. The curve at the bottom shows the emission spectrum of CPBH only under this condition. | ||
Fluorescence quenching can be a direct consequence of any process which decreases the fluorescence intensity, such as excited state reactions, energy transfers, ground-state complex formation and collision between excited molecules and other molecules.44 As introduced in the introduction, static quenching is primarily caused by the ground-state complex formation process, while dynamic quenching is largely caused by the collision between the chromophore and the quencher.1–4,14–17
In order to study the quenching process systematically and to distinguish the possible quenching mechanism, fluorescence quenching tests were performed at different temperatures to distinguish the quenching mechanism by the temperature dependence.45 The Stern–Volmer equation (eqn 1) was applied to obtain the quenching constant from the data of fluorescence spectra at each temperature:
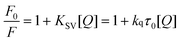 | (1) |
Herein, F0 and F are the fluorescence intensities without and with CPBH, respectively. [Q] is the concentration of the quencher, CPBH here, while KSV stands for the Stern–Volmer quenching constant. τ0, which is a constant, represents the bimolecular average fluorescence lifetime and equals 10−8 s ; kq, which equals KSV/τ0, is the apparent bimolecular quenching rate constant. Specifically pointing out, for dynamic quenching, the maximum scattering collisional quenching constant of various quenchers is 2.0 × 1010 L mol−1 s−1.1–5,10,18,46,47Fig. 2 shows the plots of the Stern–Volmer equation at different temperatures, while KSV and kq obtained from the Stern–Volmer equation are presented in Table 1. It shows clearly that KSV and kq increase with increasing temperature, suggesting that the quenching process for CPBH to SA may be a dynamic quenching. However, kq for this CPBH-SA system is much larger than 2.0 × 1010 L mol−1 s−1, suggesting that the quenching process may be a static quenching. Herein, the data show some conflicts with the classic quenching mechanism, so we need more proof to verify the real quenching process. Thus we employed UV-visible absorption spectra and electrochemical experiments to give some more evidence for the actual quenching process.
![Stern–Volmer plots for the interaction of CPBH and [A] BSA and [B] HSA at different temperatures.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f2.gif) | ||
| Fig. 2 Stern–Volmer plots for the interaction of CPBH and [A] BSA and [B] HSA at different temperatures. | ||
| BSA | HSA | ||||||
|---|---|---|---|---|---|---|---|
| pH | T (K) | K SV (105 M−1) | R a | k q (1013 M−1 s−1) | K SV (105 M−1) | R b | k q (1013 M−1 s−1) |
| a R is the correlation coefficient for Stern–Volmer plots for BSA. b R is the correlation coefficient for Stern–Volmer plots for HSA. | |||||||
| 7.4 | 298 | 1.23 | 0.9998 | 1.23 | 1.01 | 0.9992 | 1.01 |
| 303 | 1.34 | 0.9999 | 1.34 | 1.12 | 0.9994 | 1.12 | |
| 308 | 1.51 | 0.9995 | 1.51 | 1.27 | 0.9991 | 1.27 | |
| 313 | 1.70 | 0.9993 | 1.70 | 1.48 | 0.9994 | 1.48 | |
The UV-visible absorption spectra of SA, CPBH and the SA-CPBH system were measured, and we compared the spectra of SA and a subtracting spectrum to confirm the quenching mechanism. All the spectra are shown in Fig. 3. The absorbance of SA (curve B) changes at around both 280 nm (about 0.04 in absorption) and 220 nm (about 0.1 in absorption), comparing to the subtracting spectra (curve C). According to the theory mentioned in the introduction, the UV spectra of SA would have no detectable change if the quenching was a dynamic mechanism.10,14,16,17 It is the same to say, the spectra of SA and the subtracting spectra would overlay in a exact way if the quenching is a dynamic one. On the other hand, a ground-state SA–drug complex forms in the static quenching, and the UV spectrum of SA changes as a direct consequence.46 Therefore, according to the UV spectra, the fluorescence quenching of SA in our case seems to be primarily caused by complex formation between CPBH and SA.
![UV-vis absorption spectra of CPBH, SA and SA–CPBH system. [A] for CPBH–BSA system, [B] for CPBH–HSA system. c(BSA/HSA) = c(CPBH) = 10 μM](/image/article/2012/RA/c1ra00521a/c1ra00521a-f3.gif) | ||
| Fig. 3 UV-vis absorption spectra of CPBH, SA and SA–CPBH system. [A] for CPBH–BSA system, [B] for CPBH–HSA system. c(BSA/HSA) = c(CPBH) = 10 μM | ||
All the evidences above were based on the optical spectra. In case of errors caused by the optical technology, we also used electrochemistry to provide some information for the adsorption of CPBH by SA. In the electrochemical experiments, we employed a dry adsorption method to immobilize the SA molecules onto the surface of a Au electrode. This method had been proven to be useful for this research system in our previous work.10,42 Comparing the electrochemical impedance spectroscopy of a bare Au electrode (Fig. SI 1[A], curve A†) with the SA-modified Au electrode (Fig SI 1[A] curve B†), we can see that the Rct value of the bare Au electrode is very small, calculated to be 366 Ω, while the Rct value of SA-modified Au electrode is large, calculated to be 3426 Ω. Based on the comparison, we could say that SA is actually immobilized onto the surface of the Au electrode, therefore blocking the transportation of electrons. In our testing system, we only injected CPBH to the electrochemical system, and CPBH itself possessed only weak adhesive ability onto the surface of the bare Au electrode (Fig. SI 1[B],† and see the discussion at the end of this section), so the change in the Rct value is indicative of surface binding of CPBH and blocking.
In Fig. 4, Rct increases after the addition of CPBH and Rct increases with the increasing concentration of CPBH, compared to SA barely. These phenomena indicate that CPBH binds to SAvia the formation of a complex on the surface of the Au electrode which increases the resistance. Rct shows a positive correlation with the concentration of CPBH, suggesting there may be a constant binding rate between CPBH and SA in this situation. As a conclusion from the electrochemical experiments, the interactions between CPBH and SA primarily arise from the complex formation instead of collision.
![Electrochemical impedance spectroscopy (EIS) of CPBH and SA system. Various volumes of CPBH interact with [A] BSA and [B] HSA. The concentration of CPBH for A–J (μM): 0, 11.5, 23.1, 46.2, 69.3, 92.4, 115.5, 138.6, 161.7, 184.8.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f4.gif) | ||
| Fig. 4 Electrochemical impedance spectroscopy (EIS) of CPBH and SA system. Various volumes of CPBH interact with [A] BSA and [B] HSA. The concentration of CPBH for A–J (μM): 0, 11.5, 23.1, 46.2, 69.3, 92.4, 115.5, 138.6, 161.7, 184.8. | ||
The experiments of UV and electrochemistry detailed above confirm that the quenching process is primarily due to complex formation, while molecular collision could be neglected in the concentration range studied. Accordingly, the dominant mechanism of interaction between CPBH and SA is a complex formation process.
As to the unexpected and adverse temperature dependence, we think it may be an untypical static quenching and could be explained by Arrhenius' theory. As well accepted, for dynamic quenching, kq is limited by the number of excited molecules, diffusion and the probability of collision, limiting it to values less than 2.0 × 1010 L mol−1 s−1.1–5,10,18,46,47 However, there is no such limitation for static quenching. The viscosity of the solvent decreases as the temperature increases. As a result, the molecules of CPBH move faster, so they have a higher probability to collide with SA. Hence the dynamic quenching contributes more to the total quenching, and KSV decreases as the temperature increases.10,47 On the other hand, the value of KSV must increase with increasing temperature according to Arrhenius' theory. If the extent of the increase caused by the rising temperature is larger than decrease of collision, overall there will be an increase in KSV upon increasing temperature.
According to Arrhenius' theory, the rate constant k is a function of temperature, so the temperature has a crucial impact towards the rate constant k. The higher the temperature is, the higher the rate constant is. Meanwhile the activation energy Ea is related to the rate constant k. If we change the temperature to the same degree, the bigger Ea a reaction has, the greater the change to the rate constant k is.
The activation energy of the quenching process can be calculated according to the Arrhenius equation (eqn 2):
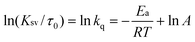 | (2) |
k q, which is equal to KSV/τ0, is the apparent bimolecular quenching rate constant; Ea is the activation energy of the quenching reaction; A is the pre-exponential factor. From the slopes of lnkqvs. 1/T plots, the value of Ea can be obtained. Fig. 5 shows a good linear relationship. The values of Ea are 16.3 and 19.5 kJ mol−1 for BSA and HSA, respectively, higher than those for many other reactions between organic ligands and serum albumin.48,49 This indicates that the impact of temperature on the value of KSV is significant. Thus, the fluorescence quenching is still a static quenching, though untypical to some extent, and CPBH forms a complex with SA in the ground state. The reason for the adverse temperature dependence for this binding process will be discussed below with the docking results.
![Arrhenius plots of CPBH and [A] BSA and [B] HSA system.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f5.gif) | ||
| Fig. 5 Arrhenius plots of CPBH and [A] BSA and [B] HSA system. | ||
Proved to be a complex formation process, a modified Stern–Volmer equation (eqn 3) is applied to calculate the affinity constant Ka of the binding between CPBH and SA.
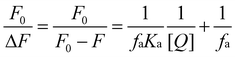
| (3) |
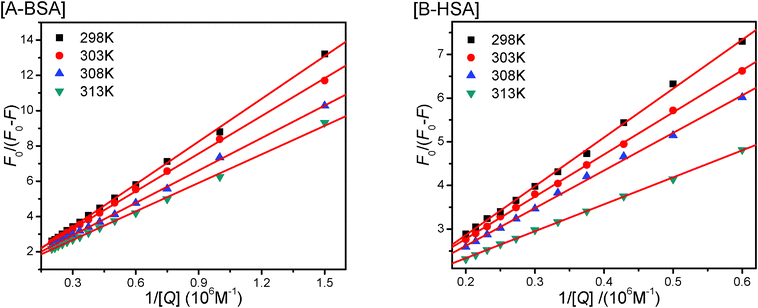 | ||
| Fig. 6 The modified Stern–Volmer plots of CPBH and SA at different temperatures. | ||
| BSA | HSA | |||||||
|---|---|---|---|---|---|---|---|---|
| T (K) | K a (105 M−1) | R | ΔG (kJ mol−1) | K a (105 M−1) | R | ΔG (kJ mol−1) | ||
| 298 | 1.22 | 0.9996 | −29.0 |
ΔH = 24.1 kJ mol−1
ΔS = 178.3 J K−1 mol−1 |
0.56 | 0.9993 | −27.1 |
ΔH = 56.5 kJ mol−1
ΔS = 280.5 J K−1 mol−1 |
| 303 | 1.52 | 0.9997 | −30.1 | 0.88 | 0.9997 | −28.7 | ||
| 308 | 1.75 | 0.9997 | −30.9 | 1.05 | 0.9990 | −29.6 | ||
| 313 | 1.95 | 0.9991 | −31.7 | 1.78 | 0.9997 | −31.5 | ||
In order to verify the data obtained by fluorescence, we have calculated the affinity constant from the data of EIS according to the Langmuir isotherm model (eqn 4):50
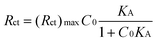 | (4) |
A plot of C0/Rct as a function of C0 demonstrates a linear relation from which the affinity constant KA can be calculated, which is shown in Fig. 7. The value of KA, calculated from the intercept of the line at C0 = 0, equals to 3.64 × 105 and 2.87 × 105 L mol−1 for BSA and HSA, respectively. This value is a little higher than that measured via the fluorescence titration. Maybe, this difference is caused by the interactions between CPBH and SA in which the binding could occur at all possible sites. In the EIS experiments, any possible binding will increase the Rct of the system. In the fluorescence titration, only the binding occurring mainly around the tryptophan residue is disclosed. Therefore, we think the values of KA measured by EIS are consistent with those measured from the fluorescence spectra.
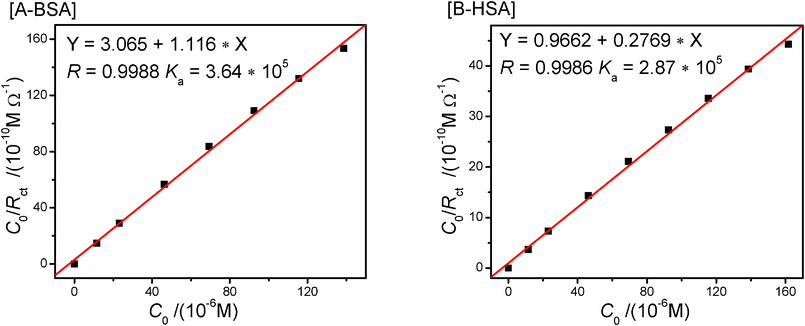 | ||
| Fig. 7 Plots of the Langmuir isotherm for CPBH and SA system. | ||
Considering of disturbance the adsorption of CPBH by the bare Au electrode, we tested the EIS of CPBH using a unmodified Au electrode. As showed in Fig. SI 1[B],† the Rct increases with the addition of CPBH, indicating that the CPBH is adsorbed to the surface of the electrode. We calculated the affinity constant of this system according the Langmuir isotherm theory (the inset in Fig. SI 1[B]†), and found it to be 0.99 × 104 L mol−1, with an acceptable linear relationship. Comparing this to the value for CPBH-SA, the adsorption binding strength of CPBH onto the surface of electrode is so weak, and could be ignored in the range of experimental error. So the adsorption of CPBH to the bare Au electrode here doesn't interfere with the binding of CPBH to the immobilized SA. And the change of Rct corresponds to the binding behavior of CPBH to SA at the surface of electrode.
3.2 Binding properties of CPBH-SA
The values of ΔH and ΔS can be estimated via the van't Hoff equation (eqn 5):
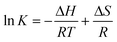 | (5) |
where K is analogous to the effective quenching constant Ka at the same temperature and R is the gas constant. It can be seen from Fig. 8 that there is a good linear relationship between ln K and 1/T, which means that the enthalpy change (ΔH) is constant in the range of temperature discussed. The enthalpy change (ΔH) can be measured from the slope of the van't Hoff plot while the entropy change (ΔS) can be obtained from the intercept. All the detailed data are listed in Table 2. As summarized by Ross and Subramanian,44,45,47 here ΔH > 0 and ΔS > 0 together suggest that the driving force is mainly a typical hydrophobic interaction, and hydrogen bonds and van der Waals forces are not dominant.
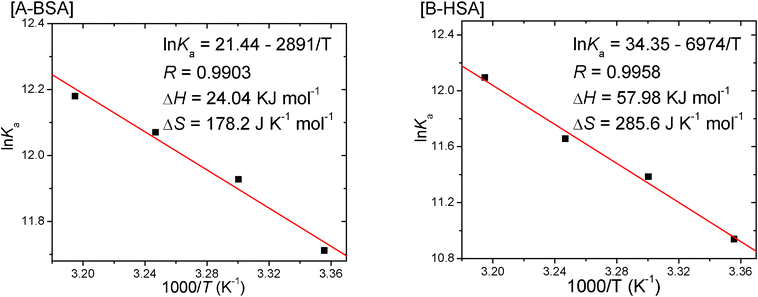 | ||
| Fig. 8 Plots of the van't Hoff equation of CPBH and SA at different temperatures. | ||
The value of ΔG at different temperatures can be calculated by eqn 6:
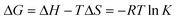 | (6) |
As the quenching is a spontaneous process, the Gibbs free energy of this quenching process is negative (ΔG < 0). Accordingly, the entropy change of the system is absolutely positive (ΔS > 0). Therefore, CPBH-induced quenching of SA fluorescence is an entropy-driven spontaneous process based on the analysis above, however, the change of enthalpy is detrimental to the spontaneous process. The enthalpy change is positive, which means that the energy level of the CPBH-SA system goes up. This change will make the complex of CPBH-SA unstable. However, the large positive entropy change neutralizes the situation caused by the unfavorable enthalpy change and stabilizes the complex. This large and favorable entropy change may be caused by micro-environmental change in the binding process.
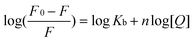
| (7) |
| pH | T (K) | BSA | HSA | ||
|---|---|---|---|---|---|
| n | R | n | R | ||
| 7.4 | 298 | 0.98 | 0.9996 | 1.13 | 0.9998 |
| 303 | 0.98 | 0.9999 | 1.07 | 0.9994 | |
| 308 | 0.94 | 0.9996 | 0.99 | 0.9995 | |
| 313 | 0.94 | 0.9993 | 0.93 | 0.9988 | |
:1. Then the fluorescence spectra were measured in Fig. 9 and analyzed.
![Spectra of CPBH–BSA/HSA system in the presence of site marker warfarin[A] and ibuprofen[B]. T = 303 K, λex = 295 nm; c(warfarin) = c(ibuprofen) = c(BSA) = c(HSA) = 2 μM; c(CPBH)/μM, A–P: 0; 0.33; 0.67; 1.00; 1.33; 1.67; 2.00; 2.33; 2.67; 3.00; 3.33; 3.67; 4.00; 4.33; 4.67; 5.00. The curve at the bottom shows the emission spectrum of warfarin [A] and ibuprofen [B] only under this condition. The insert plots correspond to the Stern–Volmer plots.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f9.gif) | ||
| Fig. 9 Spectra of CPBH–BSA/HSA system in the presence of site marker warfarin[A] and ibuprofen[B]. T = 303 K, λex = 295 nm; c(warfarin) = c(ibuprofen) = c(BSA) = c(HSA) = 2 μM; c(CPBH)/μM, A–P: 0; 0.33; 0.67; 1.00; 1.33; 1.67; 2.00; 2.33; 2.67; 3.00; 3.33; 3.67; 4.00; 4.33; 4.67; 5.00. The curve at the bottom shows the emission spectrum of warfarin [A] and ibuprofen [B] only under this condition. The insert plots correspond to the Stern–Volmer plots. | ||
Fig. 9 [A]-BSA and Fig. 9[A]-HSA show that the addition of warfarin slightly changes the fluorescence spectra of SA, characteristic of a weak red shift in the maximum emission wavelength and lower fluorescence intensity compared to the absence of warfarin. These indicate that warfarin has been bound to SA. Further injection of CPBH, which can compete against the binding of warfarin and SA at the same sites, produces fluorescence spectra shown in Fig. 9[A]-BSA and Fig. 9[A]-HSA for the color curves. This is supported by the gradual intensity decrease after the addition of ibuprofen. It shows the same phenomenon with the site marker ibuprofen, and can be seen clearly in Fig. 9[B]-BSA and Fig. 9[B]-HSA.
Taking CPBH-BSA as a example, to compare the effect of warfarin and ibuprofen on the binding of CPBH to BSA, the binding constant are calculated and compared. The binding constant of just the CPBH-BSA system is about 1.840 × 105 L mol−1 at 298 K, while those in the presence of warfarin and ibuprofen are 0.854 × 105 L mol−1 and 1.563 × 105 L mol−1, respectively. The binding constant in the presence of warfarin is much smaller than that in its absence, while that for ibuprofen is almost the same as without it. These reveal that the competition between CPBH and warfarin or ibuprofen is quite likely, and CPBH mainly competes with warfarin in sub-domain IIA (Sudlow's site I). This indicates that CPBH mainly binds to BSA in Sudlow's site I. A similar conclusion, CPBH binds to HSA in site I, can be easily obtained from Fig. 9[A]-HSA and [B]-HSA, proving the similarity of BSA and HSA as well. Herein, confirmed by the site competitive replacement experiments, we can be confident that CPBH binds to both BSA and HSA in sub-domain IIA (Sudlow's site I).
A representative isothermal calorimetric titration profile of 0.06 mM CPBH with 0.01 mM HSA in PBS at pH 7.4 and 25 °C is shown in Fig. 10 [A]. CPBH shows an approximately 1:1 binding stoichiometry (n = 1.17) towards HSA with a binding constant of 3.45 × 105 L mol−1, which suggests a strong and specific interaction. These data are identical to those obtained by fluorescence and EIS. The binding process is an endothermic process with a reaction enthalpy of 106.8 kJ mol−1. The corresponding standard free energy of binding (ΔG) and the entropy contribution (ΔS) are −31.6 kJ mol−1 and 464.4 J K−1 mol−1, respectively. For the CPBH-BSA system, the results are similar in Fig. 10 [B]: the binding constant is 4.46 × 105 L mol−1, while the binding number, n = 0.98, is very close to 1, indicating a strong and independent binding site. The enthalpy change (ΔH) is 85.5 kJ mol−1, so the corresponding standard free energy of binding (ΔG) and the entropy contribution (ΔS) are −32.2 kJ mol−1 and 395 J K−1 mol−1, respectively. These results here are very similar to the data obtained by fluorescence.
![Thermogram of CPBH binding to HSA[A] and BSA[B]. [A] HSA-CPBH system. The concentration of HSA is 10 μM in 15 mM PBS, pH = 7.4, whereas the concentration of CPBH is 60 μM. [B] BSA-CPBH, the concentration of BSA is 20 μM in 15 mM PBS, pH = 7.4, while 160 μM CPBH serves as the titrant. Each peak in the upper panel represents a single injection of the CPBH solution into the protein solution. The panels at the bottom show the plots of the amount of heat liberated per injection as a function of the molar ratio of the CPBH to the protein. The solid line represents the simulation results from the data of the titration using the NanoAnalyze software purchased from TA Instruments, USA.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f10.gif) | ||
| Fig. 10 Thermogram of CPBH binding to HSA[A] and BSA[B]. [A] HSA-CPBH system. The concentration of HSA is 10 μM in 15 mM PBS, pH = 7.4, whereas the concentration of CPBH is 60 μM. [B] BSA-CPBH, the concentration of BSA is 20 μM in 15 mM PBS, pH = 7.4, while 160 μM CPBH serves as the titrant. Each peak in the upper panel represents a single injection of the CPBH solution into the protein solution. The panels at the bottom show the plots of the amount of heat liberated per injection as a function of the molar ratio of the CPBH to the protein. The solid line represents the simulation results from the data of the titration using the NanoAnalyze software purchased from TA Instruments, USA. | ||
Together with the discussion above, the binding reaction is entropically driven, aiding CPBH-SA complex formation, whereas the positive enthalpy change component is unfavorable, destabilising the CPBH-SA complex. The strong endothermicity observed in the binding process might suggest the involvement of dehydration and the breaking of hydrogen bonds in the protein caused by the binding of CPBH. Furthermore, this makes the peptide more flexible and changes the microenvironment of the amino acid residues to a higher potential level. Nevertheless, the desired entropy change may be caused in two ways. Firstly, the peptide is more flexible. Secondly, when CPBH binds to SA in the binding site, it will force out the water molecules in and around the cavities together with the water in the gaps between amino acid residues. Overall, the complex formation of the CPBH-SA system is an entropy-driven spontaneous process.
Herein, the binding properties detected by calorimetry are accordant with all the results obtained through other methods. That is to say, the calorimetry experiments we conducted are suitable to study this CPBH-SA system, and the conclusion is supported by spectroscopy and electrochemistry.
3.3 Modeling study of the unusual static quenching
As discussed above, we know that CPBH binds to BSA and HSA at an equivalent binding site, Sudlow's site I. However, there is nothing about the accurate configuration for CPBH in the binding site, and the interaction force at the atomic level. And most importantly, the reason of the unusual static quenching remains unclear from the discussion above. In order to study the structural cause of the unusual quenching mechanism and the exact conformation of CPBH at the binding sites, to identify the precise binding sites on SA and to study the interactions between CPBH and SA systematically, we have run a docking program to simulate the binding mode between CPBH and SA.The BSA model obtained shows the similarity with HSA in structure and the equivalent binding sites based on the homology similarity of 88% in their amino acid sequence.10,55 As a result of the docking study, the exact binding site in SA is shown in Fig. 11 [A] and [B]. This docking reveals the most probable binding site and position in the protein. The detailed docking results are shown in Fig. 11 [C] and [D]. CPBH binds to both BSA and HSA at Sudlow's site I, similar to warfarin. This result is identical to the site competitive replacement experiments. The tryptophan residue has been involved in the binding sites and is at a reasonable distance from the molecule of CPBH. Thus the energy can be transported from the excited tryptophan to the CPBH by the dipole–dipole interaction, thus quenching the fluorescence emission.
![The docking results of CPBH and SA system. The binding site of CPBH in BSA[A] and HSA[B]. The configuration of CPBH in the binding site of BSA[C] and HSA[D] and the residues lying around the binding sites. CPBH is shown in space-filling model with the hydrogen bonds shown. Involved tryptophan residues are in the red circle. [E] The surface model of CPBH in the binding site of the BSA. The surface of the BSA is a red line, while Arg218 is shown in green. [F] The surface model of CPBH in the binding site of the HSA. The surface of the HSA is a red line, while Lys195 and Asp451 are in green.](/image/article/2012/RA/c1ra00521a/c1ra00521a-f11.gif) | ||
| Fig. 11 The docking results of CPBH and SA system. The binding site of CPBH in BSA[A] and HSA[B]. The configuration of CPBH in the binding site of BSA[C] and HSA[D] and the residues lying around the binding sites. CPBH is shown in space-filling model with the hydrogen bonds shown. Involved tryptophan residues are in the red circle. [E] The surface model of CPBH in the binding site of the BSA. The surface of the BSA is a red line, while Arg218 is shown in green. [F] The surface model of CPBH in the binding site of the HSA. The surface of the HSA is a red line, while Lys195 and Asp451 are in green. | ||
For the CPBH-BSA system, the amino acid residues involved in this binding site are comprised of Ile207, Ser215, Arg218, Gln219, Arg220, Trp237, Leu242, Leu261, His265, Arg280, Ala284, Ile313 and Ala314, referring to Fig. 11[C] and Fig. SI 2.† There are more than 10 amino acid residues lying around CPBH, and the interaction between CPBH and BSA is strong. Three hydrogen bonds have been observed in the CPBH-BSA complex. Atom O11 in CPBH makes a hydrogen bond with HH11 in Arg218, and another hydrogen bond with HH22 in Arg222. Atom N2 in CPBH forms a H-bond with the HE21 in the residue of Gln219. As shown in Fig. SI 2[B],† the pyridine group inserts into a hydrophilic loop made by His265, Arg280, Arg222, Arg220, Ser215 and Gln219, while the 4-chlorophenyl group stretches into a hydrophobic loop formed by Ala284, Ala314, Ile313, Leu261, Trp237 and Ile207, as shown clearly in Fig. SI 2[A].† The formation of hydrogen bonds reduces the energy of the CPBH-BSA complex and set CPBH in a ideal 3D space position to adpot the shape of the pocket of site I. The hydrophobic interaction determines the binding strength of CPBH to BSA. The hydrophilic force helps to stabilize the complex of CPBH-BSA.
For CPBH-HSA system, shown in Fig. 11[D] and Fig. SI 3,† the amino acid residues involved in this binding site are comprised of Phe211, Trp214, Ala215, Leu219, Arg222, Phe223, Leu238, Val241, Leu260, Ala261, Ile264 and Ile290. The CPBH forms three hydrogen bonds with the residues of Arg222: N2 in CPBH forms a H-bond with HE in Arg222, meanwhile O11 makes two H-bonds with HE and HH21 in Arg222, see Fig. SI 3.† As shown in Fig. SI 3,† the pyridine group in CPBH inserts into a hydrophobic loop formed by Ile290, Phe223, Ala261, Ile264, Leu260, Leu219 (Fig. SI 3[A]†). The 4-chlorophenyl group stretches into a hydrophobic pocket made by Leu238, Val241, Ala215, Trp214 and Phe211(Fig. SI 3[C]†). The two hydrophobic loops mentioned above are buried in pockets created by hydrophilic residues (Fig. SI 3[B] and [D]†). So the primary driving force of the interaction between CPBH and HSA is a hydrophobic interaction, while the hydrogen bonds and van der Waals forces play a minor part.
The results discussed here suggests that the equivalent binding site of CPBH on BSA and HSA is site I, and the main force in the interaction is a hydrophobic interaction whereas the van der Waals forces help to enhance the binding. Finally the hydrogen bonds reduce the energy of the CPBH-SA complex, and help to stabilize the complex of CPBH-SA.
As to the structural cause of the unusual static quenching mechanism, this can be explained by Fig. 11[E] and [F]. As we can see in the figures, the binding pockets are deeply buried in the body of SA, not on the surface of the protein. As a result, there should be a channel for CPBH to enter the binding pocket. CPBH has to pass through the channel formed by the amino acid residues outside the binding sites before settling at the proper position in the protein. We find that there is no other way to enter the pocket but the pocket mouth, which can be seen clearly in Fig. 11[E] and [F]. However, the one and only entrance into the binding pocket, the pocket mouth here, is closed by some amino acid residues that lying just in the middle of the ‘porch’, acting like a door (shown in green Fig. 11[E] and [F], Fig. SI 2[C] and Fig. SI 3[E]†). For BSA, the porch is made up of Glu315, Arg241 and Tyr173, and then Arg218 is working as the door panel (Fig. 11[E] and Fig. SI 2[C]†). If the door panel moves aside, the pathway is clear, and the molecule of CPBH, in the shape of a rod, can get through and fit into the binding site. Moving this residue out of its natural position requires a higher energy, which explains the large activation energy of the binding process discussed in the above sections. For HSA-CPBH, the entrance pathway is surrounded by His288, Glu292, Trp214 and His242. The porch door is made up of two amino acid residues, Lys195 and Asp451(Fig. 11[F] and Fig. SI 3[E]†). These two amino acid residues are not linked together. Each of them can move aside to make the porch door open. The molecules of CPBH must push the residues of Lys195 and Asp451 to get though the pocket mouth. This process is against the binding process and actually increases the Ea requirement of the binding process. The door of BSA is shut by only one residue, while there are two residues lying in the middle of the pathway of HSA, so the Ea requirement for HSA is larger than that of BSA. These large activation energy requirements discussed here result in the unusual temperature dependence in the fluorescence quenching discussed earlier.
Herein, we can conclude that all results obtained from the docking study are identical with the experiment results. So it is reasonably to conclude that the 3D structure of BSA is reasonable and suitable for docking studies and a deep insight of the structure-quenching mechanism relationship between CPBH and SA has been successively explored. Thus in the docking studies, we have demonstrated the differences and similarities between BSA and HSA, and the differences and similarities in the binding process between CPBH and BSA/HSA successfully.
4. Conclusion
As discussed above, CPBH, a novel acylhydrazone derivative, can be strongly adsorbed to both HSA and BSA in an equivalent and independent binding site in Sudlow's site I, forming a 1:1 complex. The unusual static quenching was caused by the large Ea requirement in the binding process, which is a direct consequence of the overcoming of the residues outside the pocket mouth which work as barriers. The dominant driving force of this interaction was a hydrophobic interaction, whereas hydrogen bonding helps CPBH to adopt the shape of the pocket in the binding site.
The results constituted a helpful guide in attempting to unravel the mechanism of CPBH-protein interactions, as well as the CPBH analogy. The results directly revealed that the adaptability between the structure of the protein and the structure of ligands has a great impact in the interaction between protein and ligands.
Acknowledgements
We gratefully acknowledge financial support from the 973 program from the Chinese Ministry of Science and Technology (2011CB939600), the National Natural Science Foundation of China (21077081, 20921062), Fundamental Research Funds for Central Universities (1103005) and support by the “Fundamental Research Funds for the Central Universities” (No. 201120302020005).References
- Y. Hu, Y. Liu, T. Sun, A. Bai, J. Lu and Z. Pi, Int. J. Biol. Macromol., 2006, 39, 280–285 CrossRef CAS.
- C. Yan, P. Mei, Z. Guan and Y. Liu, Chin. J. Chem., 2007, 25, 1085–1089 CrossRef CAS.
- C. Yang, Y. Liu, D. Zheng, J. Zhu and J. Dai, J. Photochem. Photobiol., A, 2007, 188, 51–55 CrossRef CAS.
- Y. Hu, Y. Liu and X. Xiao, Biomacromolecules, 2009, 10, 517–521 CrossRef CAS.
- D. V. Suresh, H. G. Mahesha, A. G. Appu Rao and K. Srinivasan, Biopolymers, 2007, 86, 265–275 CrossRef CAS.
- J. Tian, J. Liu, W. He, Z. Hu, X. Yao and X. Chen, Biomacromolecules, 2004, 5, 1956–1961 CrossRef CAS.
- D. Shcharbin, E. Pedziwiatr, L. Chonco, J. F. Bermejo-Martín, P. Ortega, F. J. Mata, R. Eritja, R. Gómez, B. Klajnert, M. Bryszewska and M. A. Muñoz-Fernandezand, Biomacromolecules, 2007, 8, 2059–2062 CrossRef CAS.
- B. Zhou, Z. Zhang, Y. Zhang, R. Li, Q. Xiao, Y. Liu and Z. Li, J. Pharm. Sci., 2009, 98, 105–113 CrossRef CAS.
- R. Beauchemin, C. N. N'soukpoé-Kossi, T. J. Thomas, T. R. Thomas, Carpentier and H. A. Tajmir-Riahi, Biomacromolecules, 2007, 8, 3177–3183 CrossRef CAS.
- F. Tian, F. Jiang, X. Han, C. Xiang, Y. Ge, J. Li, Y. Zhang, R. Li, X. Ding and Y. Liu, J. Phys. Chem. B, 2010, 114, 14842–14853 CrossRef CAS.
- R. Beauchemin, C. N. N'soukpoé-Kossi, T. J. Thomas, T. R. Carpentier and H. A. Tajmir-Riahi, Biomacromolecules, 2007, 8, 3177–3183 CrossRef CAS.
- U. S. Mote, S. L. Bhattar and S. R. Patil, Luminescence, 2010, 25, 1–8 CAS.
- X. Pan, J. Liu and D. J. Zhang, J. Colloid Interface Sci., 2010, 345, 442–447 CrossRef CAS.
- J. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd edn., Plenum, New York, 1999 Search PubMed.
- M. K. Johansson and R. M. Cook, Chem.–Eur. J., 2003, 9, 3466–3471 CrossRef CAS.
- J. N. Demas and J. W. Addington, J. Am. Chem. Soc., 1974, 96, 3663–3664 CrossRef CAS.
- H. Chen, S. S. Ahsan, M. B. Santiago-Berrios, H. D. Abruña and W. W. Webb, J. Am. Chem. Soc., 2010, 132, 7244–7245 CrossRef CAS.
- J. Hu, Y. Ou-Yang, C. Dai, Y. Liu and X. Xiao, Biomacromolecules, 2010, 11, 106–112 CrossRef.
- R. Sivakumar, S. Naveenraj and S. J. Anandan, J. Lumin., 2011, 131, 2195–2201 CrossRef CAS.
- A. C. Vaiana, H. Neuweiler, A. Schulz, J. Wolfrum, M. Sauer and J. C. Smith, J. Am. Chem. Soc., 2003, 125, 14564–14572 CrossRef CAS.
- P. Cheng, Debbie. Silvester, G. Wang, G. Kalyuzhny, A. Douglas and R. W. Murray, J. Phys. Chem. B, 2006, 110, 4637–4644 CrossRef CAS.
- Y. Hu, H. Yu, J. Dong, X. Yang and Y. Liu, Spectrochim. Acta, Part A, 2006, 65, 987–991 CrossRef.
- Y. Zhang, J. Dai, X. Zhang, X. Yang and Y. Liu, J. Mol. Struct., 2008, 888, 152–159 CrossRef CAS.
- N. Akbay, Z. Seferoglu and E. Gokoglu, Current Analytical Chemistry, 2010, 10, 334–340 Search PubMed.
- Z. Qi, Y. Zhang, F. Liao, Y. Ou-Yang, Y. Liu and X. Yang, J. Pharm. Biomed. Anal., 2008, 46, 699–706 CrossRef CAS.
- S. Rollas and Ş. G. Küçükgüzel, Molecules, 2007, 12, 1910–1939 CrossRef CAS.
- D. S. Kalinowski, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2008, 51, 331–344 CrossRef CAS.
- D. A. Green, W. E. Antholine, S. J. Wong, D. R. Richardson and C. R. Chitambar, Clin. Cancer. Res., 2001, 7, 3574–3579 CAS.
- J. Andreas, F. J. Carsten, B. Wolfgang, P. Rene and E. Dieter, Tetrahedron, 2002, 58, 2253–2329 CrossRef.
- E. Dieter, D. Sylvie, S. Daniel and H. Audrey, Chem.–Eur. J., 2007, 13, 3942–3949 CrossRef.
- R. Lygaitis, V. Getautisc and J. V. Grazulevicius, Chem. Soc. Rev., 2008, 37, 770–788 RSC.
- K. Malmos, M. Dong, S. Pillai, P. Kingshott, F. Besenbacher, S. U. Pedersen and K. Daasbjerg, J. Am. Chem. Soc., 2009, 131, 4928–4936 CrossRef CAS.
- S. G. Kucukguzel, E. E. Oruc, S. Rollas, F. Sahin and A. Ozbek, Eur. J. Med. Chem., 2002, 37, 197–206 CrossRef CAS.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS.
- D. R. Richardson, D. S. Kalinowski, S. Lau, P. J. Jansson and D. B. Lovejoy, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 702–717 CrossRef CAS.
- D. B. Lovejoy and D. R. Richardson, Blood, 2002, 100, 666–676 CrossRef CAS.
- D. S. Kalinowski, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2007, 50, 6212–6225 CrossRef CAS.
- D. M. Himmel, S. G. Sarafianos, S. Dharmasena, M. M. Hossain, K. McCoy-Simandle, T. Ilina, A. D. Clark, J. L. Knight, J. G. Julias, P. K. Clark, K. Krogh-Jespersen, R. M. Levy, S. H. Hughes, M. A. Parniak and E. A. C. S. Arnold, Chem. Biol., 2006, 1, 702–712 CAS.
- J. R. Dimmock, S. C. Vashishtha and J. P. Stables, Eur. J. Med. Chem., 2000, 35, 241–248 CrossRef CAS.
- L. Bukowski, M. Janowiec, Z. Zwolska-Kwiek and Z. Andrzejczyk, Pharmazie, 1999, 54, 651–654 CAS.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS.
- R. Li, F. Jiang, Q. Xiao, J. Li, X. Liu, Q. Yu, Y. Liu and C. Zeng, Nanotechnology, 2010, 21, 475102, DOI:10.1088/0957-4484/21/47/475102.
- B. Valeur. Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2001 Search PubMed.
- Y. Zhang, X. Xiang, P. Mei, J. Dai, L. Zhang and Y. Liu, Spectrochim. Acta, Part A, 2009, 72, 907–914 CrossRef.
- Y. Hu, Y. Liu, J. Wang, X. Xiao and S. Qu, J. Pharm. Biomed. Anal., 2004, 36, 915–919 CrossRef CAS.
- B. K. Sahoo, K. S. Ghosh and S. Dasgupta, Biopolymers, 2008, 91, 108–119 CrossRef.
- J. Chen, X. Y. Jiang, X. Q. Chen and Y. Chen, J. Mol. Struct., 2008, 876, 121–126 CrossRef CAS.
- X. Han, P. Mei, Y. Liu, Q. Xiao, F. Jiang and R. Li, Spectrochim. Acta, Part A, 2009, 74, 781–787 CrossRef.
- A. Barik, B. Mishra, A. Kunwar and K. I. Priyadarsini, Chem. Phys. Lett., 2007, 436, 239–243 CrossRef CAS.
- D. Li, X. Zou, Q. Shen and S. Dong, Electrochem. Commun., 2007, 9, 191–196 CrossRef CAS.
- S. Bjelic and I. Jelesarov, J. Mol. Recognit., 2008, 21, 289–311 CrossRef CAS.
- X. Zhou, Q. Sun, R. Manjunatha Kini and J. Sivaraman, Protein Sci., 2008, 17, 1798–1804 CrossRef CAS.
- Y. Liang, Acta Biochim. Biophys. Sin., 2008, 40, 565–576 CrossRef CAS.
- O. Okhrimenko and I. Jelesarov, J. Mol. Recognit., 2008, 21, 1–19 CrossRef CAS.
- S. N. Khan, B. Islam, R. Yennamalli, A. Sultan, N. Subbarao and A. U. Khan, Eur. J. Pharm. Sci., 2008, 35, 371–382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The design principle for CPBH, the control of EIS, the detailed illustration of modeling result, the 1H NMR, MS, IR, UV-visible spectra of CPBH. See DOI: 10.1039/c1ra00521a |
| This journal is © The Royal Society of Chemistry 2012 |