Chrome yellow in nineteenth century art: historic reconstructions of an artists' pigment†
Vanessa
Otero
ab,
Leslie
Carlyle
ab,
Márcia
Vilarigues
ac and
Maria J.
Melo
*ab
aREQUIMTE-CQFB, Faculty of Sciences and Technology, New University of Lisbon, 2829-516, Monte da Caparica, Portugal. E-mail: mjm@dq.fct.unl.pt; Fax: (+351) 212948322; Tel: (+351) 212948322
bDepartment of Conservation and Restoration, Faculty of Sciences and Technology, New University of Lisbon, 2829-516, Monte da Caparica, Portugal
cVICARTE, Faculty of Sciences and Technology, New University of Lisbon, 2829-516, Monte da Caparica, Portugal
First published on 22nd December 2011
Abstract
To understand the reported degradation of chrome yellows, popular with artists since their introduction in the 19th century, it is necessary to understand the pigment formulation as produced at that time. Chromium-based pigments such as lead chromate (PbCrO4, chrome yellow) or zinc chromate (K2O·4ZnCrO4·3H2O, zinc yellow), as used by Van Gogh and Seurat, currently exhibit substantial darkening in paintings such as “Sunflowers” or “A Sunday on La Grande Jatte-1884”. Winsor & Newton (W&N), one of the leading artists' colourmen of the time, has made its recipe archive available. Access to their extensive chrome yellow pigment formulations prompted our research on the stability of these pigments by reconstructing their processes of manufacture. The colorants obtained were compared with contemporary tube paints from W&N as well as with samples from paintings by Amadeo de Souza-Cardoso (1887–1918) an influential modernist Portuguese painter. Good correlation between all three sources was found.
1. Introduction
Detailed knowledge of the materials used by artists is essential to reveal their techniques and to place their works in context, as well as to ensure effective conservation and authentication procedures.1 Artist's oil paints commercially available today do not reflect 19th century technology, both in relation to the oil binder as well as in the formulations for the colorants. In order to understand the degradation mechanisms that are in play in such a complex material as a 19th century oil painting it is necessary to have historically accurate reference materials.2,3 These are used in the laboratory to observe the natural evolution of an oil paint, and also to simulate by accelerated ageing experiments with a limited number of variables, the aging of these paint systems.Following the discovery of chromium in 1797 by Vauquelin and his synthesis of lead chromate in 1809,4 chrome yellow (PbCrO4) made its entry to the 19th century artists' palette. Its commercial production started in England between 1814 and 1816 with Dr Bollman and in the USA and France a few years later.5 Although modern chrome yellow manufacture includes encapsulation and additives not found in the 19th century,6 industrial production is still essentially based on the same steps that were already developed by Vauquelin: a solution of a soluble lead salt (nitrate or acetate) is added to a chromate (CrO42−) or dichromate solution (Cr2O72−). In solution these Cr6+ species are in an acid–base equilibrium (Fig. 1), which is easily shifted towards the chromate anion, by adding cations such as Ba2+, Pb2+ and Ag+ that lead to the formation of insoluble chromates (instead of soluble dichromates).7 In our experimental conditions, following the steps to achieve chrome yellow detailed in W&N's archive, only Cr6+ ions were present and no redox chemistry was observed.
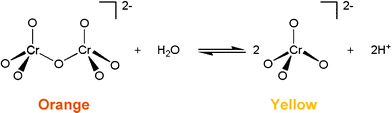 | ||
| Fig. 1 Acid base equilibrium established in the range 1 < pH < 11, pKa ≈ 6.4 between chromate ion (CrO42−) and dichromate ion (Cr2O72−).7 | ||
Our process for producing chrome yellow was based on four recipes dating from 1846 to 1858 from the W&N database (Researchers’ Edition). The Researchers’ Edition combines a computer-based indexing system with digitalised page by page images from 85 handwritten books detailing manufacturing practices and recipes for 19th century artists' materials.8,9 These recipes include the pigment as well as additional materials (see below). As will be demonstrated, it was important to synthesise the pigment ourselves for a variety of reasons, not the least is that modern sources of this pigment include particle coatings. These protective treatments were first developed in the 1950s using inert inorganic (and more recently organic) coatings in order to improve the pigment's stability or its application.6 In fact, concerns regarding chrome yellow's stability were recorded in artists' manuals throughout the 19th century (e.g. Mérimée (1839) and Vibert (1892)).10,11 The darkening of chromate pigments has also been a subject of concern for conservators.12,13 However the pigment's observed darkening behaviour in oil is inconsistent: this has been reported in works by Van Gogh,13 but in the works of Amadeo that we have studied, chrome yellows appear to be unchanged.12,14,15 Recently, systematic work on the degradation of chrome-based pigments, involving artificially ageing studies and the analysis of microsamples from artworks has concluded that the main mechanism of degradation results in reduced chrome species, namely Cr3+ oxo compounds, as for example Cr2O312,16 and, rather unexpectedly, green viridian, Cr2O3·2H2O.17 Studies carried out by Casadio et al.12 on the degradation of zinc yellow also suggested the possibility of the formation of dichromate species. Moreover, the formation of sulfite and/or sulfate anions was judged a key factor in chrome yellow degradation.13,17 These researchers consider that all these species are formed in the topmost surface (3–5 μm) or upper layers of the paint, and are likely poorly crystalline, therefore their characterisation is a challenging issue. In these studies no full explanation was offered for the data obtained in the aging experiments conducted on historic tube paint samples, which to a certain extent, appears to reveal different degradation pathways, for example when darkening is observed in some samples but not in others.13,17 This is not surprising given the diversity of chrome yellow formulations, the complexity of the ageing process in an oil matrix, together with the fact that the original paintings where this pigment is found have likely been subject to restoration procedures, in particular moisture and solvent exposure (in the latter case through varnish removal). In the work discussed here, the knowledge of chrome yellows is based on direct characterisation of the pigment and other associated materials from microsamples of artworks and old oil paint tubes. Both cases represent 100 years of chemical evolution within an oil matrix. Creating pigment samples according to representative historic recipes offers the opportunity to fully characterise the powdered pigment, before it is mixed with oil, and before the aging process has introduced change. The next step, to create oil paint with the pigment using highly characterised linseed oil3 allows significant progress to be made in our understanding of the interaction with the binder and to compare this with reported degradation mechanisms and the state of the materials in naturally aged paints. With this methodology we expect to be able to predict with more accuracy the evolution of a particular colour in oil and the main factors at play. Full characterisation of the pigment in oil was beyond the scope of this paper, which concentrates on the reconstructions of the historic pigment formulations.
2. Results and discussion
2.1. Historic reconstructions of W&N chrome yellows
Four recipes were selected from a set of 79 (see Experimental 3.1) based on the amount of detail provided in the recipe (the recipes with the most information) and whether the recipe represented a relatively common formulation (from the range present). Notes accompanying these four recipes described the pigments formed as “rich” and “bright”. Basically, in these recipes, potassium dichromate was added to lead nitrate (Pb(NO3)2) or lead subacetate (Pb(Ac)2·2Pb(OH)2), in the presence of sodium carbonate (Fig. 2). Other materials were added to this solution, including plaster of Paris (CaSO4·1/2H2O), barites (BaSO4) or chalk (CaCO3). In the W&N recipes, the plaster of Paris had been previously converted into gypsum, by stirring it in water for two hours. Therefore in our reconstructions this material was added as gypsum (CaSO4·2H2O), see Experimental for more details.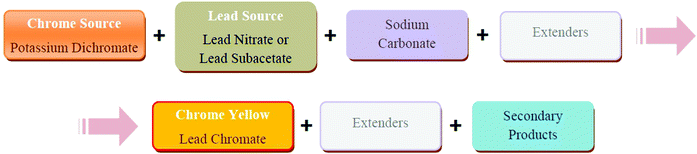 | ||
| Fig. 2 Fundamental steps in the W&N chrome yellow process. | ||
All products of the recipes were characterised by microRaman, XRD, SEM, microFTIR and colorimetry (Lab* coordinates ranging from 76,23,71 to 85,30,89). MicroRaman and microFTIR were preferred to Raman and FTIR because when comparing with samples from original paintings these are two fundamental and complementary techniques, which allow full characterisation of chrome yellow chromophores and additives in very small samples. With microFTIR it is possible to gather quantitative information on the proportions of pigment, additives and binder. Initially the recipes were broken down into what we considered the fundamental steps to investigate variables: Step 1 reacting the different lead compounds with the chrome solutions; Step 2 reacting the chrome and lead sources in the presence of each of the possible additional materials (e.g.gypsum, barites, or chalk). Then finally in Step 3 each full recipe was reconstructed (described in the text as the “complete recipe” or process and coded P1 through to P4). The reactions that were carried out in (1) and (2) enabled us to better understand the role played by each of the additional materials and to offer a rationale for the reactions and products obtained (namely unexpected by-products as will be described below). For all four “complete recipes” that are summarised in Table 1 and Fig. 3, the final pH was neutral, leading to the formation of lead chromate as the main pigment. Interestingly, for all the processes carried out within Step 2, pigment precipitation was always observed and some of the products match with those found in samples from historic paintings and paints.
| Process/recipe name | Reagents | pH | ||||
|---|---|---|---|---|---|---|
| P1 Best Middle Chrome | Potassium dichromate | Sodium carbonate | Calcite | Lead nitrate | — | 6.70 |
| P2 Super Lemon Chrome | Sodium sulfate | Lead nitrate | Gypsum | 7.31 | ||
| P3 Best Middle Chrome | Sulfuric acid | Barites | Lead subacetate | 6.46 | ||
| P4 Best Yellow Chrome | Sulfuric acid | Lead subacetate | Gypsum | 7.60 | ||
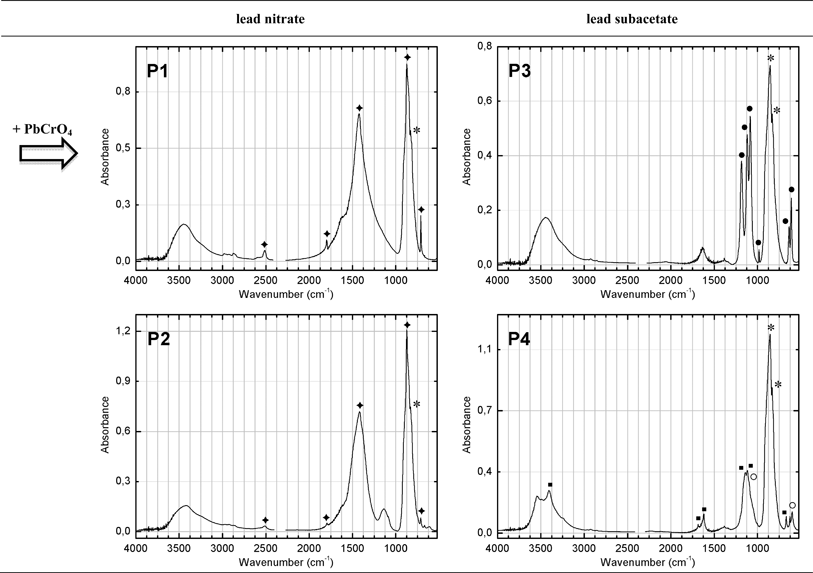 | ||
| Fig. 3 Infrared spectrum for the pigments obtained, in which the main absorptions are assigned to: (*) PbCrO4, (◆) CaCO3, (●) BaSO4, (■) CaSO4·2H2O and (○) Pb(Cr,S)O4. | ||
In two of the processes (P1 and P2), lead nitrate was mixed with a buffer solution of sodium carbonate and potassium dichromate. In P1, calcium carbonate was also added and acts both as an extender and for pH control. In P2, sodium sulfate was mixed in the chrome solution and after the addition of the lead nitrate, gypsum was introduced. Both P1 and P2 result in a formulation where lead chromate is found together with calcium carbonate, in a proportion of ca. 50%, taking as representative the spectra depicted in Fig. 3. Whereas calcium carbonate is a deliberate addition in P1, in the case of P2 it is present as a by-product, resulting from the conversion of a sodium into a calcium carbonate (solubility product, Ksp = 3.36 × 10−9) at the expenses of gypsum (Ksp = 3.14 × 10−5).
The other two processes (P3 and P4) used the same source for chromate as in P1 and P2, but lead was introduced as lead subacetate; sulfuric acid was added to the chrome solution before the lead subacetate. For P3, the extender barium sulfate was introduced prior to the lead subacetate solution, resulting in a formulation where lead chromate is mixed with barium sulfate (Fig. 3). In P4 the extender gypsum was added at the end. In this latter case, together with gypsum and lead chromate, mixed-crystals of lead chromate and lead sulfate (Pb(Cr,S)O4) were also obtained (Fig. 3). Both infrared and Raman spectroscopy are able to fingerprint these mixed (Cr,S) chromophores. In the infrared spectrum, identifying the content of sulfate ions in the crystalline structure is straightforward due to the intensity of the SO42− vibrational bands, which are shifted from those of pure lead sulfate. In addition, the CrO42− asymmetric stretching profile also changes, shifting to higher wavenumbers. By Raman spectroscopy, differences are found in the intensity of the bands in the CrO42− bending region and in the appearance of the SO42− symmetric stretching band at 968 cm−1. This is in agreement with XRD results where it was possible to observe that as the content of sulfate ions increases, the shift from the crocoite XRD pattern is larger (see ESI†).18,19 The mixed-crystals usually display a paler yellow hue when compared with the pure lead chromates. On the other hand, bright yellows are obtained in the presence of extenders in the formulation such as calcium carbonate or barium sulfate. Calcium carbonate may also improve the mechanical properties of the paint film and it is possible that it may play a protective role as a pH buffer. Fig. 3 and 4 depict the infrared spectra and SEM images of the final products from the reconstructions that will be next compared with samples from old oil paint tubes and oil paintings. In terms of morphology, SEM-BSE images show the characteristic rod-like particles of lead chromate, however the various production methods resulted in different particle sizes ranging from 1 μm to 3 μm in length and 0.1 μm to 1 μm in width. In general, these particles are longer than those reported by Burnstock et al.20, where lead chromate rod-like particles with a maximum length of 1 μm were observed.
 | ||
| Fig. 4 SEM images for the pigments obtained with P1 to P4 recipes. Note the presence of calcium carbonate visible as a grey mass in P1 (deliberate addition) and P2 (by-product of pigment synthesis). | ||
It is also noteworthy that, in P1, lead carbonate was also formed, in a small proportion, as a by-product, at the expense of sodium carbonate and in the absence of sulfuric acid (Ksp(PbCO3) = 7.4 × 10−14, Ksp(PbCrO4) = 1.8 × 10−14).
In the “complete” reconstructions described above, we were able to investigate the role of individual reagents as well as the chemistry of the full formulations. We will now discuss unexpected but very interesting results obtained within Steps 2 and 3. “Pure” lead chromate was obtained when the lead source was combined with the chromate buffered solution, as described in Step 1 above. The production of red/orange phoenicochroite, Pb2CrO5, was observed when lead subacetate and potassium dichromate were made to react at a basic pH; i.e., when H2SO4 was not added in P3 or P4 or when Na2CO3 was substituted by NaOH in P1 and P2. Pb2CrO5, characterised by XRD and detected by FTIR, may be formed together with lead chromate or as a major compound. Presumably to avoid this reaction and to obtain the yellow form of the pigment, W&N, in P3 and P4 syntheses, added sulfuric acid to the chrome solution prior to the addition of the lead source. Phoenicochroite was also used as an artists' pigment, and was known as chrome orange or basic lead chromate (see Burnstock et al.20, where it was identified in paint samples dating from 1870).
2.2. Comparison with historic paint samples
Two examples from historic oil paints were selected to compare with the reconstructed pigments: a W&N chrome yellow oil paint tube that had belonged to Columbano Bordalo Pinheiro (1857–1929) and a Lefranc chrome yellow oil paint tube belonging to Amadeo de Souza-Cardoso (1887–1918).Using hand-ground oil paints made with our synthesised chrome yellow pigments it was possible to determine that an unaged oil matrix does not interfere in the fingerprint region for the chrome yellow formulations. Therefore, comparison between the synthesised pigments and the historic oil paint samples could be made directly using spectra from our powdered pigments (Fig. 3, and see the Experimental for more details). A very good match was found for Columbano's W&N chrome yellow oil with the product from P1 (Fig. 5a), in which all the main features in the infrared molecular fingerprint from the tube paint can clearly be observed. Calcium carbonate is identified unequivocally through its intense and broad band at 1422 cm−1 and sharp absorption at 875 cm−1. This absorption overlaps with the CrO42− asymmetric stretching band at 853 cm−1 (shoulders at 831 and 820 cm−1), which is characteristic of lead chromate. This suggests that the chrome yellow pigment used in Columbano's oil paint was manufactured by a process similar to P1 and P2, where the chrome source is added to a lead nitrate solution. The higher content of calcium carbonate may be explained as a result of mixing different proportions of the reagents.
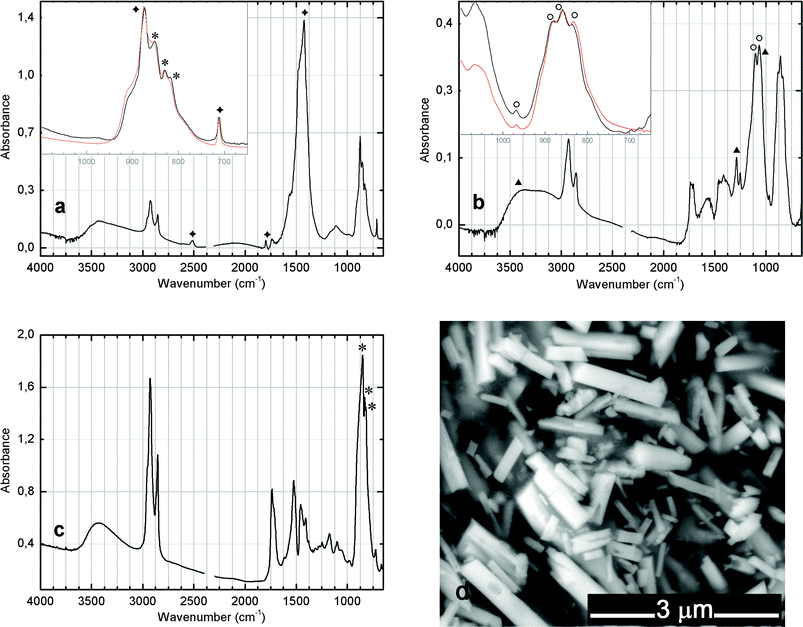 | ||
| Fig. 5 (a) Infrared spectrum of Columbano's oil paint tube (Chrome deep, W&N). In the inset, comparison with P1 pigment (orange line) for 1100–650 cm−1; (b) Infrared spectrum of Mucha micro-sample. In the inset, comparison with P2 pigment (orange line) for 1100–650 cm−1. (*) PbCrO4, (◆) CaCO3, (○) Pb(Cr,S)O4 and (▲) Cr2O3·2H2O (viridian); (c) Infrared spectrum of Amadeo's oil paint tube (Jaune de Chrome foncé – Lefranc) and (d) respective SEM image in BSE mode. | ||
The Lefranc yellow tube paint is a pure pigment in an aged oil matrix (Fig. 5c). It appears most similar to what we obtained by mixing the lead with the chrome source at an acidic or neutral pH as described in Step 1. The infrared spectra for the Lefranc sample is characteristic of a “pure” lead chromate displaying a well defined CrO42− asymmetric stretching profile, triply degenerate, with an intense band at 851 cm−1 (shoulders at 830 and 818 cm−1). This is in agreement with the SEM-BSE image, where only lead chromate rod-like particles are observed (compare with Fig. 4).
In addition, a light green micro-sample from Amadeo's painting Mucha (1915–16) was analysed (see Fig. 5b and 6). A high content of mixed-crystals of lead chromate and lead sulfate was identified, together with the green pigment viridian (characteristic bands at 3630–2630, 1288 and 1064 cm−1). This spectrum is a very good match with P2 product.
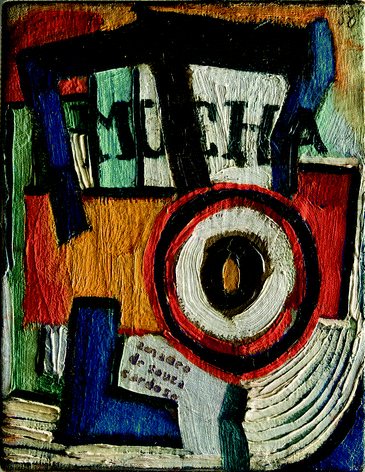 | ||
| Fig. 6 Amadeo's painting: Mucha (1915–1916) taken in ranking light. | ||
2.3. Impact on current degradation models for chrome based compounds
The results obtained have shown that mixed compounds such as CaSO4·nH2O (characterised by FTIR and XRD) or phoenicochroite can result during the original synthesis of the pigment. The fact that dichromate was used as a starting material must also be taken into account when analysing degradation phenomena directly on original painting samples. In the publications referred to above, dichromate ion was proposed as a degradation product based on a comparison of electron energy loss (EEL) reference spectra of powders, which did not contain phoenicochroite.12,16 In another publication, gypsum which was presented as playing a key role in the chrome yellow degradation, was thought to be produced during ageing, however our work shows that it can also have been present in the original pigment formulation.2.4. Impact on the interpretation of additions to artists’ paint
Two by-products identified in the synthesis of chrome yellow pigments, calcium carbonate and lead carbonate, would, before this work, likely have been interpreted in historic oil paint samples as deliberate additions during paint manufacture (thought to modify paint rheological properties, texture or hue, or, in the case of calcium carbonate, as an extender). The reconstructions using actual historical formulations demonstrate that these materials can be present as a result of the conditions during pigment manufacture.3. Experimental
3.1. Winsor & Newton recipes for chrome yellow
Seventy-nine records were found in the Researchers' Edition of the W&N archive database pertaining to chrome yellow manufacture. The majority of the 79 records were dated between 1846 and 1865, indicating that the company's experiments, development and commercial manufacture of this pigment was concentrated within this period. As W&N no longer manufactures lead chromate-based pigments (their modern chrome yellow hue is based on organic compounds21), there were no recipe restrictions in the Researchers' Edition. In the four W&N recipes selected, chrome yellow was obtained by adding one of the lead solutions (nitrate or subacetate) to a solution of potassium dichromate. Additional ingredients called for were chalk, barites and gypsum. “Soda” was present in all four recipes. In order to obtain lead chromate, “soda” had to be sodium carbonate. If the alternate interpretation for soda was used (sodium hydroxide), then phoenicochroite was the main colorant produced (a chrome orange rather than a chrome yellow).A total of 33 pigments were produced, and the synthetic methods used for the “complete recipes” are described in Table 2. The unique recipe codes of the four recipes are: P1P245AL01 for P1 (Best Middle Chrome); P1P433AL01 for P2 (Super Lemon Chrome); P1P192AL01 for P3 (Best Middle Chrome) and P2P009AL01 for P4 (Best Yellow Chrome).
| Lead solution | Chrome solution | Pigment production | |
|---|---|---|---|
| P1 | 9.4 ml boiling H2O | 50 ml H2O | Chrome solution |
| + | + | + | |
| 1.5 ml HNO3 | 1.325 g K2Cr2O7 in 12.5 ml boiling H2O | Sift 1.649 g CaCO3 | |
| + | + | + | |
| Sift 2.650 g PbO | 0.795 g Na2CO3 in 12.5 ml boiling H2O | 9.7 ml lead solution | |
| Boil for 1 h, stirring | |||
| P2 | 9.4 ml boiling H2O | 50 ml of H2O | Chrome solution |
| + | + | + | |
| 2.8 ml HNO3 | 1.325 g K2Cr2O7 in 12.5 ml boiling H2O | 1.855 g Na2SO4 in 12.5 ml boiling H2O | |
| + | + | + | |
| Sift 3.869 g PbO | 1.484 g Na2CO3 in 12.5 ml boiling H2O | 9.7 ml lead solution (stirring for 1 h) | |
| Boil for 1 h, stirring | + | ||
| 1.855 g CaSO4·2H2O sifted into 74 ml H2O and stirred for 2 h | |||
| P3 | 13.6 ml boiling H2O | 50 ml of H2O | Chrome solution |
| + | + | + | |
| 1.178 g Pb(Ac)2·3H2O | 1.325 g K2Cr2O7 in 12.5 ml boiling H2O | 0.3 ml H2SO4 (drop by drop) | |
| + | + | + | |
| Sift 1.412 g PbO | 0.795 g Na2CO3 in 12.5 ml boiling H2O | Sift 1.237 g BaSO4 | |
| Boil for 2 h, stirring | + | ||
| 9.4 ml lead solution (stirring for 20 min) | |||
| P4 | 20 ml boiling water | 50 ml of water | Chrome solution |
| + | + | + | |
| 3.092 g Pb(Ac)2·3H2O | 1.325 g K2Cr2O7 in 12.5 ml boiling H2O | 0.6 ml H2SO4 (drop by drop) | |
| + | + | + | |
| Sift 2.760 g PbO | 1.104 g Na2CO3 in 12.5 ml boiling H2O | Add lead solution till neutralised | |
| Boil for 1 h, stirring | + | ||
| 1.546 g CaSO4·2H2O sifted into 20 ml H2O and stirred for 2 h |
3.2. Apparatus
To fully characterise each pigment a multi-analytical approach was used. X-Ray Diffraction (XRD) provided the crystal structure; Scanning Electron Microscopy (SEM) characterised the pigment morphology. Energy-Dispersive X-Ray Fluorescence spectroscopy (EDXRF) gave the elemental composition of the pigments and enabled semi-quantification of the ratio of pigment to other additives. The fingerprinting techniques of microRaman spectroscopy and microFourier Transform Infrared spectroscopy (FTIR) provided pigment characterisation at the molecular level.XRD were obtained at the Materials Department of FCT/UNL and SEM, within the Portuguese microscopy network REM, at CEMUP-Centro de Materiais University of Porto. All the other analysis were carried out at the DCR-FCT/UNL.
FTIR is able to fingerprint the chrome yellow pigment structure as confirmed by comparison and correlation with XRD data. Also, in case studies samples Raman may also be a very useful technique, but in this paper we choose to present mainly FTIR combined with SEM-EDX because, together, they enable for a complete characterisation and semi-quantification of pigments and secondary products.
X-Ray powder diffraction (XRD) patterns were recorded on a Rigaku Dmax III-C 3 kW diffractometer, using monochromatised Cu-Kα radiation at 40 kV and 20 mA settings in the 10° < 2θ < 80° range at a scanning speed of 2° per minute.
SEM images were obtained using an FEI Quanta 400 FEG ESEM, which uses a Schottky emitter field emission gun, operating at low vacuum conditions and at 15 kV, equipped with an EDAX Genesis X4M detector. Images were acquired using secondary (SE) and backscattered (BSE) electron detectors.
X-Ray fluorescence spectra were obtained with an ArtTAX spectrometer of Intax GmbH, with a molybdenum (Mo) anode, Xflash detector refrigerated by the Peltier effect (Sidrift), with a mobile arm. The spatial resolution is 70 mm. The experimental parameters used were: 40 kV of voltage, 300 μA of intensity and 200 s of acquisition time.
MicroRaman microscopy was carried out using a Labram 300 Jobin Yvon spectrometer, equipped with a He-Ne laser of 17 mW power operating at 632.8 nm. Spectra were recorded as an extended scan. The laser beam was focused with a 50× Olympus objective lens. The laser power at the surface of the samples was varied with the aid of a set of neutral density filters (optical densities 0.3, 0.6, 1).
Infrared analyses were carried out using a Nicolet Nexus spectrophotometer coupled to a Continuμm microscope (32× objective) with an MCT detector cooled by liquid nitrogen. The pigments were prepared as KBr pellets, and spectra were collected in transmission mode, with a resolution of 4 cm−1 and 64 scans. For the microsamples of oil paint tubes and oil paintings, spectra were obtained in transmission mode, 4000–650 cm−1, with a resolution of 4 cm−1 and 128 scans, using a Thermodiamond anvil compression cell. The spectra are shown here as acquired, without corrections or any further manipulations, except for the occasional removal of the CO2 absorption at ca. 2300–2400 cm−1.
Conclusions
The W&N archive page-image database provides efficient access to a unique source of information on 19th century artists' materials and their commercial preparation. The investigation on the chemical methods for producing 19th century chrome yellow pigments and the characterisation of the products has provided a much deeper understanding, not only of the chemistry of the materials, but also the individual steps taken to achieve the end products. In this way, this work contributes significant new knowledge concerning historical pigment manufacture as well as an understanding of “the manufacturer's rationale for producing a specific range”.20 It also contributes to the history of chemistry.Moreover, mapping the chemistry of the pigment by recreating historic formulations, including the detection of materials present as by-products brings new insights to studies on the degradation of 19th century chrome yellow paints and opens new perspectives for the design of artificially ageing experiments to investigate pigment and pigment/oil stability. Compounds that were interpreted as degradation products in samples from original 19th century paintings may be now re-evaluated with the possibility that they could have been present in the original pigment formulation. Given that the darkening of chrome-based pigments is affecting such iconic paintings as Seurat's “A Sunday on La Grande Jatte-1884” and Van Gogh's “Sunflowers” any new information regarding the degradation mechanisms for chrome yellow pigments is indeed an important advance.
This demonstration of the value of the W&N archive page-image database for reproducing historic pigment manufacturing methods constitutes a turning point in the study of the molecular ageing of oil paintings. We have begun with the investigation of chrome yellow pigments due to the problems they pose for some key paintings and the work already done by others to elucidate their degradation. However we plan to extend our research using the archive database to other pigments and, in future, to explore reconstructions of 19th century oil paint formulations.
Acknowledgements
The authors are grateful to Winsor & Newton, ColArt Fine Art & Graphics Ltd., in particular Emma Pearce and Ian Garrett for making the archive project possible, and most recently for the kind assistance of Paul Robinson. The W&N archive project was funded by the Netherlands Institute for Scientific Research (NWO) as part of the De Mayerne Programme and in the UK, by a Resource Enhancement Grant from the Arts and Humanities Research Council (AHRC). The Portuguese Science Foundation, FCT-MCTES, is acknowledged for funding the project PTDC/EAT-EAT/113612/2009 “Crossing borders: History, materials and techniques of Portuguese painters from 1850–1918” and Vanessa Otero's PhD Grant, SFRH/BD/74574/2010. Special thanks to Ana Carneiro for her valuable help with the 19th century chemical nomenclature and to João Pedro Veiga for his help in the pigment analysis by X-ray diffraction.References
- M. J. Melo and A. Claro, Acc. Chem. Res., 2010, 43, 857–866 CrossRef CAS.
- J. V. Weerd, A. V. Loon and J. J. Boon, Studies in conservation, 2005, 50, 3–22 Search PubMed.
- L. Carlyle, Hart Report. De Mayerne Programme. Technical Report, 2005 Search PubMed.
- H. Kühn and M. Curran, Artists' Pigments: a handbook of their history and characteristics, vol. 1, 1986, pp. 187–200 and 208–211 Search PubMed.
- R. D. Harley, Artists' Pigments c. 1600–1835, 2001, pp. 100–102 Search PubMed.
- G. Buxbaum, Industrial Inorganic Pigments, 1998, pp. 58 and 116–119 Search PubMed.
- F. A. Cotton and W. Geoffrey, Advanced Inorganic Chemistry, 1999, pp. 736–752 Search PubMed.
- M. Clarke and L. Carlyle, Preprints of ICOM-CC 14th Triennial Meeting, 2005, 24–29 Search PubMed.
- M. Clarke, Studies in Conservation, 2009, 54, 160–169 Search PubMed.
- J. G. Vibert, The Science of Painting, Trans. (from 8th revised edition) by Percy Young, 1892 Search PubMed.
- J. F. L. Mérimée, The art of painting in oil, and in fresco; Being a history of the various processes and materials employed, from its discovery, by Hubert and John van Eyck, to the present time, Trans. W. B. Sarsfield Taylor, 1839 Search PubMed.
- F. Casadio, S. Xie, S. C. Rukes, B. Myers, K. A. Gray, R. Warta and I. Fiedler, Anal. Bioanal. Chem., 2011, 399, 2909–2920 CrossRef CAS.
- L. Monico, G. Snickt, K. Janssens, W. Noft, C. Miliani, J. Verbeeck, H. Tian, H. Tan, J. Dik, M. Radepont and M. Cotte, Anal. Chem., 2011, 83, 1224–1231 CrossRef CAS.
- J. Kirby, K. Stoner, A. Roy, A. Burnstock, R. Grout and R. White, National Gallery Tech. Bull., 2003, 24, 4–37 Search PubMed.
- M. Vilarigues, M. J. Melo, S. Baboet al., Catálogo Raisonné, Amadeo de Souza-Cardoso – Pintura, 2008, pp. 81–104 Search PubMed.
- L. Zanella, F. Casadio, S. Xie, K. A. Gray, R. Warta, Q. Ma and J. Gaillard, J. Anal. At. Spectrom., 2011, 26, 1090–1097 RSC.
- L. Monico, G. Snickt, K. Janssens, W. Noft, C. Miliani, J. Verbeeck, H. Tian, H. Tan, J. Dik, M. Radepont and M. Cotte, Anal. Chem., 2011, 83, 1214–1223 CrossRef CAS.
- B. F. Mentzen, A. Latrach and J. Bouix, Mater. Res. Bull., 1984, 19, 549–554 CrossRef CAS.
- L. J. H. Erkens, H. Hamers, R. J. M. Hermans, E. Claeys and M. Bijnens, Surf. Coat. Int., Part B, 2001, 84, 169–176 CrossRef CAS.
- A. Burnstock, C. G. Jones and G. Cressey, Zeitschrift für Kunsttechnologie und Konservierung, 2003, 17, 74–84 Search PubMed.
- D. Pyle and E. Pearce, The Oil Colour Book: A comprehensive resource for painters, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1ra00614b |
| This journal is © The Royal Society of Chemistry 2012 |