2,5-bis(Arylethynyl)thienyl systems: Preparation and photophysical properties. Part II†
Benjamin A.
Coombs
,
Simon R.
Rutter
,
Andrés E.
Goeta‡
,
Hazel A.
Sparkes
,
Andrei S.
Batsanov
and
Andrew
Beeby
*
Department of Chemistry, University of Durham, Durham, DH1 3LE. E-mail: Andrew.beeby@durham.ac.uk; Fax: (+44)191 384 4737; Tel: (+44)191 334 2023
First published on 5th January 2012
Abstract
A series of novel thienyl aryleneethynylenes have been prepared by the Sonogashira cross-coupling of dibrominated or diiodinated thiophenes with arylacetylenes. Previously we have investigated the properties of 2,5-bis(phenylethynyl)thiophene, BPET (1), [Siddle et al., New J. Chem., 2007, 31, 841] and in this work we go on to make chemical modification of the central thienyl moiety and examine their influence on the photophysical properties of the arylethynylene thienyl system. Room and low temperature absorption and emission spectra have been recorded, along with fluorescence quantum yields and lifetimes. It is demonstrated that the 1,1-dioxides (3b & 4b) exhibit significant broadening of their fluorescence spectra relative to the other systems, which we attribute to some degree of charge transfer from the peripheral phenyl rings into the electron-deficient thiophene-1,1-dioxide moiety. Furthermore, a non-conjugated fused thiophene system (6b) displays phosphorescence at low temperature, which is rarely observed from aryleneethynylenes. DFT and TD-DFT calculations have been performed to assist understanding the observed properties.
Introduction
Aryleneethynylenes are extended π-conjugated systems with a highly linear structure. As such they have been shown to demonstrate remarkable structural, electronic and photophysical properties, lending themselves to application in sensors1,2 and as organic electronic components, including in photovoltaic cells,3 OLEDs,4,5 and as charge transfer devices i.e. molecular wires.6,7 Separately, thiophene, specifically oligo(thiophenes) and derivatives, has exhibited huge potential in the field of organic electronics; therefore it was used in one of the earliest organic field effect transistors,8 and ever since has enjoyed success as a p-type semiconductor,9–12 as well as displaying interesting photophysical properties which have lent it to applications in TFTs, LCDs and chemosensors.13–18 In an attempt to avoid unwanted by-products formed during polymerisation of thiophene and thus enhance the solubility of poly(thiophenes), Bayer AG prepared a thiophene derivative featuring an ethylenedioxy-bridge which was appended to the thiophene over the 3 and 4 positions—preventing side-chain formation and cross-linking.19 The polymer PEDOT was found to have excellent properties as an antistatic film, and is widely used in OLED devices to aid charge injection.20,21Work by Barbarella et al. has shown that chemical modification of thiophene through oxidation affords electron-deficient thiophene-based compounds that act as n-type semiconductors.22,23 Barbarella and co-workers have also demonstrated some of the interesting photophysical properties that thiophene-1,1-dioxide containing compounds can display.24–26 Materials containing fused thiophene rings have also shown great potential in the field of organic electonics.27 Their extended planarity enforces a rigidity not seen in oligo-thienyl systems. Furthermore, their packing in crystals exhibits atypical behaviour; whereas most thienyl systems pack in a herringbone pattern, fused thiophene systems invariably pack in a face-to-face pattern. This has been attributed to an S–S interaction, which is thought to stabilise the fused thiophenes relative to their non-sulfurous analogues.
Our group has long been interested in the synthesis and excited state properties of 1,4-bis(phenylethynyl)benzene, BPEB, and its derivatives.28–31 More recently alternative cyclic systems, such as anthracene and pyrazine, have been investigated.4,32, We and co-workers recently presented work on the fluorescent properties of a series of substituted BPEB-type aryleneethynylenes incorporating thiophene as the central core heterocycle (see Fig. 1).33
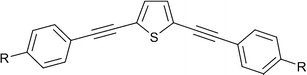 | ||
| Fig. 1 2,5-bis(arylethynyl)thiophene. | ||
This work featured a study on the effect of peripheral electron donating and withdrawing groups on the compound's photophysical properties, and showed that both electron donating groups (R = Me, OMe and NMe2) and withdrawing groups (R = CN, CF3 and CO2Me) induce a red shift in the absorption and emission spectra relative to the unsubstituted parent 2,5-bis(phenylethynyl)thiophene (R = H). This was attributed to a reduction in the HOMO–LUMO energy gap by the introduction of the two classes of substituents.
In this companion paper a series of novel thienyl arylethynylenes have been prepared and studied, each featuring modified central thiophene-based heterocycles, allowing the investigation of the effects of modification of the central ring upon the electronic structures. The modified thiophene systems discussed here are illustrated in Fig. 2. The EDOT derivative 2b, allows consideration of a more electron-rich thiophene than the parent 1, whilst oxidation of thiophene to the 1,1-dioxide enables the study of an electron-deficient system. In 4b these two functionalisations are made simultaneously. The series is completed by three isomeric fused thienothiophene systems; the fully conjugated 5b and the cross-conjugated 6b and 7b.
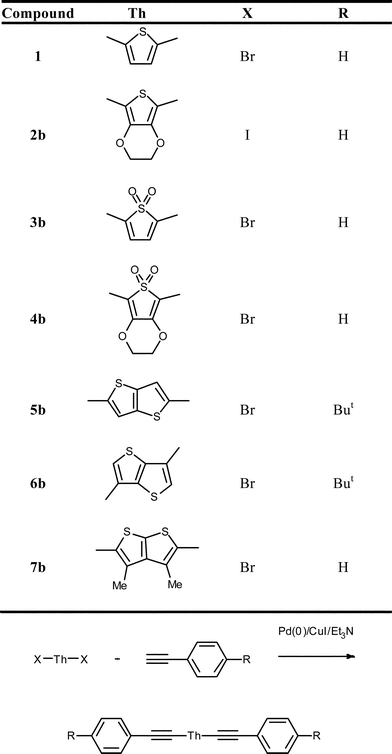 | ||
| Fig. 2 Bis(arylethynyl)thienylenes 1 & 2b–7b. | ||
Results and discussion
Synthesis
The preparation of these novel thienyl arylethynylenes was readily achieved by Sonogashira coupling of the appropriate dihalothiophene derivative, ThX2, 2a–7a with excess phenylacetylene, yielding the products 2b–7b. In some cases the 4-tertbutyl phenylacetylene was employed to improve the solubility of the product whilst having a negligible effect on the electronic structure.2,5-Diiodo-3,4-ethylenedioxythiophene (2a) was prepared following the method described by the method of Zotti et al.34N-Iodosuccinimide was reacted with 3,4-ethylenedioxy thiophene in dry DMF, affording 2a in good yield (70%). Several routes have been reported for the preparation of 2,5-dibromothiophene-1,1-dioxide. Direct oxidation of electron deficient 2,5-dibromothiophene is reported to be inefficient using conventional oxidants, requiring more forcing conditions (e.g.TFAA/H2O2, DMDO, or HOF).35–37 In the present work we employed the method described by Furukawa et al., using m-CPBA to oxidise 2,5-bis(trimethylsilyl) thiophene.38 This was subsequently brominated, affording the precursor 3a in good yield (60%). Derivative 4a was prepared by direct m-CPBA oxidation of the dibromide prepared following the method described by Tran-Vanet al.39 The anti-thienothiophene precursors (5a and 6a) were synthesised in good yields (83 and 60%, respectively) following methods adapted from those described by Iddon et al.40 The syn-thienothiophene precursor was prepared following a method described by Kirsch et al.,41 and brominated following the method reported by Campbell et al.,42 affording 2,5-dibromothieno[2,3-b]thiophene (7a) in excellent yield (90%).
The novel thienyl arylethynylenes (2b–7b) were prepared via the Sonogashira cross-coupling of the dihalo thiophene derivatives with 2.5 equivalents of phenylacetylene or 4-t-butylphenylacetylene. These coupling reactions were performed in triethylamine/THF and employed PdCl2(PPh3)2 or Pd(PPh3)4 as the catalyst, along with copper iodide as a co-catalyst, (see ESI for individual reaction amounts and conditions†). Reactions were monitored by TLC or GC-MS analysis, and final compounds were isolated using column chromatography and/or recrystallisation. Full experimental details are included in the ESI.†
Crystal structures
Crystallographic structure determination using single crystal X-ray diffraction methods has afforded molecular structures as shown in Fig. 4. This has enabled direct analysis of the bond lengths in the thienyl moieties, to help better understand the effect that chemical modification, e.g.oxidation, has on molecular structure, particularly the thienyl ring. Selected bond lengths (labelled in Fig. 3) are reported in Table 1, and full crystallographic details in Table 2.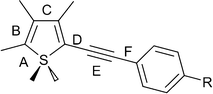 | ||
| Fig. 3 Thienyl arylethynyl motif, with bonds labelled. | ||
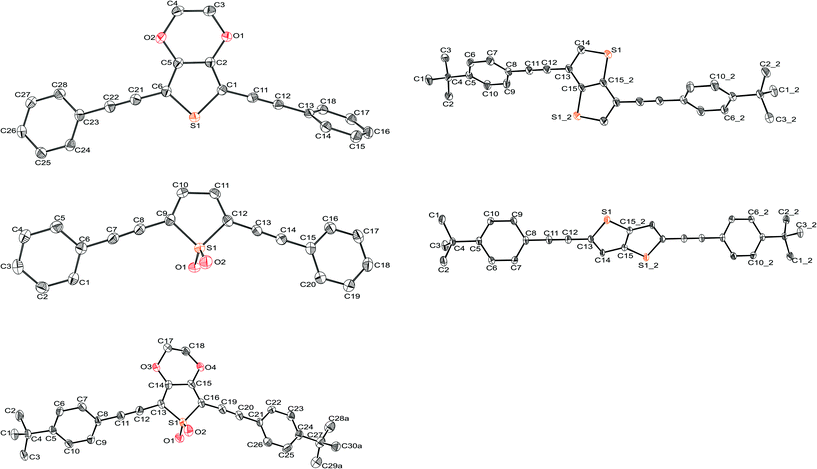 | ||
| Fig. 4 Molecular structures of several novel thienyl arylethynylenes, obtained through X-ray crystallography. | ||
| A | B | C | D | E | F | |
|---|---|---|---|---|---|---|
| 1 | 1.732(3) | 1.378(3) | 1.411(4) | 1.424(4) | 1.197(4) | 1.444(4) |
| 1.757 | 1.388 | 1.412 | 1.405 | 1.220 | 1.424 | |
| 2b | 1.737(1) | 1.372(2) | 1.418(2) | 1.415(2) | 1.199(2) | 1.435(2) |
| 1.763 | 1.385 | 1.422 | 1.399 | 1.219 | 1.421 | |
| 3b | 1.790(3) | 1.339(4) | 1.459(4) | 1.404(4) | 1.202(4) | 1.432(4) |
| 1.836 | 1.358 | 1.452 | 1.391 | 1.220 | 1.420 | |
| 4b | 1.773(3) | 1.340(3) | 1.475(3) | 1.415(4) | 1.193(3) | 1.437(4) |
| 1.824 | 1.362 | 1.466 | 1.389 | 1.220 | 1.419 |
| Compound reference | 2b | 3b-precursor | 3b | 4b | 5b | 6b |
|---|---|---|---|---|---|---|
| Formula | C22H14O2S | C10H20O2SSi2 | C20H12O2S | C30H30O4S | C30H28S2 | C30H28S2 |
| Formula Mass | 342.39 | 260.50 | 316.36 | 486.60 | 452.64 | 452.64 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Triclinic | Triclinic |
| a/Å | 13.2880(3) | 15.2943(15) | 14.1518(4) | 16.4620(8) | 6.0263(4) | 6.2158(4) |
| b/Å | 5.61860(10) | 15.4858(15) | 6.29430(10) | 7.2069(3) | 8.1335(5) | 7.6037(5) |
| c/Å | 22.5335(5) | 6.4210(6) | 18.7696(5) | 23.1514(14) | 13.0125(7) | 13.2115(10) |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 | 87.992(2) | 102.290(2) |
| β (°) | 93.0350(10) | 90.00 | 109.7410(10) | 107.172(6) | 77.1150(10) | 93.687(2) |
| γ (°) | 90.00 | 90.00 | 90.00 | 90.00 | 74.0720(10) | 97.016(2) |
| V/Å3 | 1679.99(6) | 1520.8(3) | 1573.65(7) | 2624.2(2) | 597.64(6) | 602.91(7) |
| T/K | 120(2) | 215(2) | 120(2) | 120(2) | 120(2) | 120(2) |
| Space group | P21/c | Pmmn | P21/c | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
P
|
| Z | 4 | 4 | 4 | 4 | 1 | 1 |
| Reflections collected | 18746 | 19738 | 5822 | 19062 | 8056 | 6903 |
| Unique reflections | 4792 | 2369 | 2146 | 4640 | 2949 | 2468 |
| R int | 0.0366 | 0.0349 | 0.0470 | 0.0789 | 0.0261 | 0.0374 |
| Final R1 values (I > 2σ(I)) | 0.0413 | 0.0336 | 0.0633 | 0.0508 | 0.0412 | 0.0492 |
| Final wR(F2) values (I > 2σ(I)) | 0.0895 | 0.0894 | 0.1693 | 0.0696 | 0.1122 | 0.1354 |
| Final R1 values (all data) | 0.0612 | 0.0491 | 0.0719 | 0.1261 | 0.0470 | 0.0586 |
| wR(F2) | 0.0969 | 0.1033 | 0.1815 | 0.0821 | 0.1179 | 0.1461 |
Comparing the crystallographic data for compounds 1 and 3b it is found that oxidation of the thiophene ring has a profound effect on the lengths of the bonds of the thiophene ring and those coupling it to the acetylene, A–D. We observe a dramatic lengthening of bond C, and a similarly dramatic shortening of bonds B upon oxidation of the sulfur, suggesting that in the 1,1-dioxide the thienyl ring has reduced aromaticity, in agreement with findings by Barbarella et al.22 Thus, the single C–C bond at C is longer in 3b than in 1 implying greater single bond character, whilst the double C–C bond(s) at B have shortened to adopt greater double bond character. A similar effect is seen in the EDOT derivatives 2b and 4b. Bond A is also elongated in the oxidised systems, reinforcing the fact that the sulfones are less aromatic and that the thiophene ring is more akin to a butadiene structure. Molecular structures found for 2b–6b are shown in Fig. 4. In compounds 5b and 6b it is found that the phenyl rings are significantly twisted with respect to the central thienyl group. This is attributed to packing interactions in the solid state and, based upon our experience with compounds of this type, this is not an indication that these conformations are preferred in solution. Packing of the fused thiophene systems 5b and 6b was face-to-face, in contrast to the usual herringbone-style packing seen in oligothiophenes.33
Optical spectroscopy
The optical properties of all systems were investigated by room and low temperature UV-vis absorption and fluorescence spectroscopy. The fluorescence quantum yields and lifetimes and a summary of the optical spectra for compounds 1 and 2b–7b are presented in Table 3. The room temperature absorption and fluorescence excitation profiles show good agreement (See ESI†) for all systems except compound 7b, indicating that, like the parent system, following excitation there is rapid excited state relaxation and conformational equilibration to the emissive state.28| Compound | λ max abs/nm | ε/M−1cm−1 | λ max ex/nm | λ max em/nm | Stokes shift/cm−1 | φ f/(± 10%) | τ f/(± 0.1 ns) | k f/(± 12%) |
|---|---|---|---|---|---|---|---|---|
| a From earlier work reported by Siddle et al.33 b Fluorescence lifetime could not be measured accurately. c Phosphorescence lifetime obtained at 77 K (in EPA), see text. d Fluorescence attributed to impurity. | ||||||||
| 1a | 350 | 33 000 | n/a | 382 | 2390 | 0.20 | 0.24 | 0.83 |
| 2b | 361 | 45 200 | 362 | 395 | 2380 | 0.12 | 0.13 | 0.92 |
| 3b | 415 | 21 000 | 409 | 519 | 4830 | 0.24 | 1.1 | 0.22 |
| 4b | 433 | 24 300 | 427 | 553 | 5010 | 0.02 | n/ab | n/a |
| 5b | 371 | 62 300 | 371 | 402 | 2080 | 0.43 | 0.38 | 1.13 |
| 6b | 320 | 37 900 | 320 | 341 | 1920 | 0.08 | n/ab,c | n/a |
| 7b | 322 | 42 300 | 325d | 366d | 3730 | n/ad | n/ab | n/a |
Compounds 1, 2b and 5b display similar absorption and emission profiles, with a small (∼10 nm increments) red-shifting of both the absorption and emission maxima as the central thienyl group is substituted, concomitant with an increase in conjugation. The absorption profiles display some evidence of vibronic structure, with a shoulder on both the red- and blue-edges of the maxima, whilst the emission spectra contain more pronounced vibrational fine structure. Comparison of the absorption and emission profiles of 1 and 3b, and 2b and 4b highlights the effect of the oxidation of the thiophene group; there is significant broadening of both the absorption and emission profiles and loss of vibrational features upon oxidation. This is indicative of some charge–transfer character in these compounds which feature a highly electron-deficient thiophene-1,1-dioxide core. Both 3b and 4b exhibit absorption and emission profiles devoid of any vibrational fine structure at ambient temperature, although there is a slight blue-shifted shoulder to their emission maxima. The absorption and emission maxima of 6b are the highest in energy for this series, attributable to the lack of conjugation across the whole molecule. Compound 6b displays a similar emission profile to that of its conjugated isomer 5b, although their absorption profiles are different, Fig. 5.
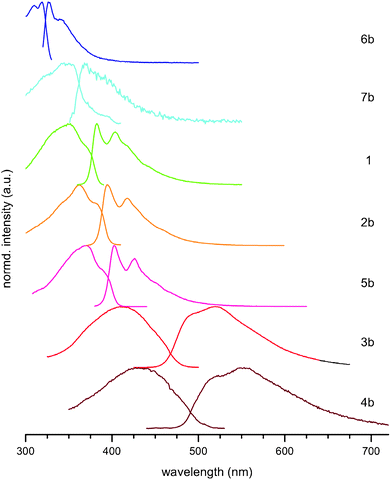 | ||
| Fig. 5 Fluorescence excitation and emission spectra for compounds 1–6b in toluene. | ||
Compound 7b shows a blue shifted absorption band and was found to be virtually non-fluorescent. Based on the poor agreement between the absorption and excitation profiles, we attribute the very weak emission to traces of fluorescent impurities and conclude that the compound itself is non-fluorescent, Φf < 0.001.
As found in BPEB, the absorption spectra obtained at room temperature for compounds 1 and 2b–6b show profiles that are not mirror images of their emission spectra.
As shown in Fig. 6 the excitation and emission spectra observed form low temperature samples exhibit enhanced vibrational fine structure, red-shifted absorption maxima and reduced Stokes shifts. The ground state of these compounds is predicted to have a shallow potential well, favouring the planar conformation over the perpendicular by 100–400 cm−1. The first excited state has a much deeper potential well, again favouring the planar conformation. Thus, at room temperature all possible conformations are present giving rise to a broadened absorption profile. Upon excitation these conformations rapidly planarise, < 50 ps, giving rise to structured emission from a narrow distribution of conformers. Cooling the samples in an EPA glass (diethylether, 2-methylbutane, alcohol in 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:2 ratio) to 77 K favours the lower energy, planar conformation, markedly changing the absorption spectra. Upon excitation there is little reorganisation in the low temperature glass and hence the emission and absorption profiles show a more mirror-image like relationship.28
:5:2 ratio) to 77 K favours the lower energy, planar conformation, markedly changing the absorption spectra. Upon excitation there is little reorganisation in the low temperature glass and hence the emission and absorption profiles show a more mirror-image like relationship.28
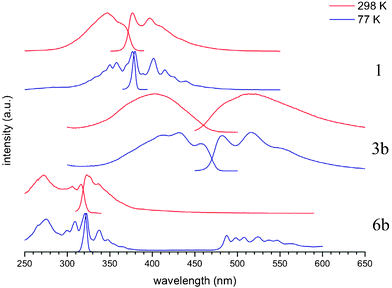 | ||
| Fig. 6 Room and low temperature (77 K) excitation and emission spectra of selected compounds. | ||
The low temperature emission spectrum of compound 6b is particularly interesting as this features an additional low energy emission band at low temperature, attributed to phosphorescence, based upon the long lifetime of 0.14 s. This is highly unusual for an aryleneethynylene derivative: phosphorescence is normally only observed when there are significant heavy atom effects, such as Pt(II) or Au(I) acetylides.43–45
Fluorescence quantum yields and lifetimes (where possible) have been measured for all systems. These are presented in Table 3. The quantum yields vary dramatically, with the non-conjugated systems 6b exhibiting relatively low quantum yields (φf = 0.08). The oxidised EDOT derivative (4b) exhibited a similarly low quantum yield, the origin for which is not known. The other systems (1, 2b–5b) feature quantum yields between 0.12 and 0.43. The absorption and emission spectra and fluorescence quantum yield and lifetimes of 3b were found to be solvent dependent. A Mataga-Lippert plot, illustrated in Fig. 7 shows a gradient of 2590 cm−1, which assuming an ellipsoidal cavity for the molecule of 50 Å3 indicate a change in dipole of 3.5 D upon excitation.
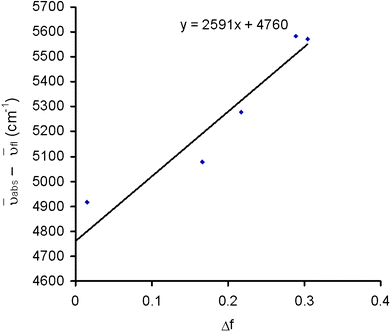 | ||
| Fig. 7 Lippert plot for compound 3b. | ||
Fluorescence lifetimes have been recorded in toluene, using the TCSPC method, recording at the emission maxima. For the systems featuring low quantum yields, lifetimes could not be measured accurately. Compounds 1, 2b and 5b featured very fast decay times, τf < 400 ps, and are calculated to have fairly consistent kf values of ∼1 ns−1. The lifetime of 3b, however, was considerably longer than these compounds, at over 1 ns. This has resulted in a significantly lower kf value, indicative of a lower oscillator strength for this process, which may reflect the charge–transfer nature of the transition.
DFT calculations
We have performed DFT and TD-DFT calculations on all systems, with geometries calculated using the B3LYP functional and 6-31+G(d) basis set.46 A co-planar arrangement of rings was found to be lowest in energy, in agreement with the systems reported by Siddle et al.33 and BPEB.28 Frequency calculations were performed on the optimised geometries of the ground electronic states for all the systems reported here to ensure that genuine minima were found. A comparison can be made between the calculated geometry and that obtained from single crystal X-ray diffraction measurements. However, not all materials could be crystallised satisfactorily. In these cases it is useful to compare the computed and observed Raman frequencies as a test of the model. Employing a wavenumber correction factor of 0.96 for all frequencies (typical for this level of theory and basis set)33 we observe good agreement between the calculated frequencies and the observed Raman spectra (see ESI†).The TD-DFT calculations allowed assignment of the S1←S0 excitation which in each case is dominated by the LUMO←HOMO transition and in all cases possesses a high oscillator strength. A plot of the computed HOMO and LUMO energies, the calculated excitation energies and observed excitation energies determined from the optical spectra are shown in Fig. 8 and Table 4.
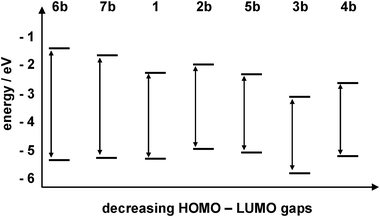 | ||
| Fig. 8 DFT-calculated HOMO–LUMO energy gaps, and the oscillator strengths calculated for the transitions. | ||
Plots of the HOMO and LUMO of the molecules 1 and 3b are illustrated in Fig. 9. As shown for the parent 1, the electron density in the HOMO and LUMO are delocalised across the entire molecule, with significant electron density on the thiophene ring, In the oxidised analogue 3b the LUMO has greater localisation over the C–SO2–C group and reduced density on the peripheral phenylethynyl groups, indicative of some degree of charge transfer character from the phenylethynyl groups to the electron deficient sulfone during the transition.
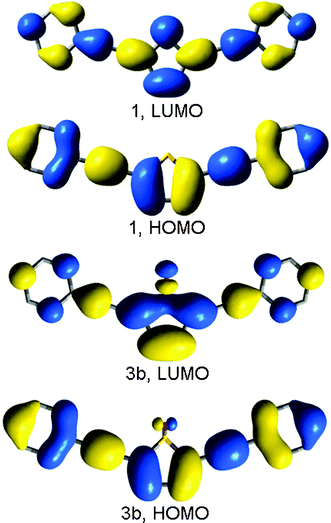 | ||
| Fig. 9 Calculated (B3LYP/6-31G*) HOMO and LUMO's for 1 and 3b. | ||
Conclusions
We have prepared and studied a range of bis(phenylethynyl)thiophene derivatives in which the central thienyl group is systematically varied. These compounds were readily synthesised by cross-coupling appropriate dibromo- and diiodinated heteroarenes under Sonogashira conditions, with arylacetylenes. The photophysical properties of these compounds have been measured and found to dramatically change through the chemical modification of the central thienyl heterocyclic core. Addition of an electron-donating ethylenedioxy bridge (2b) resulted in a slight red shift (860 cm−1) and vibrational fine structure in the emission spectrum remained. However, oxidation of the thiophene ring to generate the thiophene-1,1-dioxide resulted in a large red-shift of 6910 cm−1 and a broad absorption and emission profile for 3b, attributed to a transition with some charge transfer character, which is supported by DFT calculations. This work also presents a rare example of an arylethynylene (6b) displaying phosphorescence, allowing the triplet state to be observed.References
- J. Yang and T. Swager, J. Am. Chem. Soc., 1998, 120, 11864 CrossRef CAS.
- J. Yang and T. Swager, J. Am. Chem. Soc., 1998, 120, 5321 CrossRef CAS.
- Q. Liu, J. Mao, Z. Liu, N. Zhang, Y. Wang, L. Yang, S. Yin and Y. Chen, Nanotechnology, 2008, 19, 115601 CrossRef.
- L. Zhao, I. F. Perepichka, F. Turksoy, A. S. Batsanov, A. Beeby, K. S. Findlay and M. R. Bryce, New J. Chem., 2004, 28, 912 RSC.
- G. Mao, A. Orita, L. Fenenko, M. Yahiro, C. Adachi and J. Otera, Mater. Chem. Phys., 2009, 115, 378 CrossRef CAS.
- J. Tour, Acc. Chem. Res., 2000, 33, 791–804 CrossRef CAS.
- M. A. Reed, C. Zhou, C. J. Muller, T. P. Burgin and J. M. Tour, Science, 1997, 278, 252 CrossRef CAS.
- A. Tsumura, H. Koezuka and T. Ando, Appl. Phys. Lett., 1986, 49, 1210–1212 CrossRef CAS.
- D. Fichou, J. Nunzi and F. Charra, Adv. Mater., 1994, 6, 64 CrossRef CAS.
- D. Fichou, G. Horowitz, B. Xu and F. Garnier, Synth. Met., 1990, 39, 243 CrossRef CAS.
- F. Garnier, R. Hajlaoui, A. Yassar and P. Srivastava, Science, 1994, 265, 1684 CAS.
- P. Wu, H. Xin, F. S. Kim, G. Ren and S. A. Jenekhe, Macromolecules, 2009, 42, 8817 CrossRef CAS.
- H. Fuchigami, A. Tsumura and H. Koezuka, Appl. Phys. Lett., 1993, 63, 1372–1374 CrossRef CAS.
- A. Dodabalapur, L. Torsi and H. Katz, Science, 1995, 268, 270 CAS.
- H. Sirringhaus, R. Friend, X. Li, S. Moratti, A. Holmes and N. Feeder, Appl. Phys. Lett., 1997, 71, 3871 CrossRef CAS.
- L. Qiu, W. H. Lee, X. Wang, J. S. Kim, J. A. Lim, D. Kwak, S. Lee and K. Cho, Adv. Mater., 2009, 21, 1349 CrossRef CAS.
- H. Sirringhaus, N. Tessler and R. Friend, Science, 1998, 280, 1741 CrossRef CAS.
- T. Swager and M. Marsella, Adv. Mater., 1994, 6, 595 CrossRef CAS.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. Reynolds, Adv. Mater., 2000, 12, 481 CrossRef CAS.
- S. Oyston, C. Wang, I. Perepichka, A. Batsanov, M. Bryce, J. Ahn and M. Petty, J. Mater. Chem., 2005, 15, 5164 RSC.
- K. Kamtekar, C. Wang, S. Bettington, A. Batsanov, I. Perepichka, M. Bryce, J. Ahn, M. Rabinal and M. Petty, J. Mater. Chem., 2006, 16, 3823 RSC.
- G. Barbarella, L. Favaretto, M. Zambianchi, O. Pudova, C. Arbizzani, A. Bongini and M. Mastragostino, Adv. Mater., 1998, 10, 551 CrossRef CAS.
- G. Barbarella, L. Favaretto, G. Sotgiu, M. Zambianchi, L. Antolini, O. Pudova and A. Bongini, J. Org. Chem., 1998, 63, 5497 CrossRef CAS.
- G. Gigli, O. Inganäs, M. Anni, M. de Vittorio, R. Cingolani, G. Barbarella and L. Favaretto, Appl. Phys. Lett., 2001, 78, 1493 CrossRef CAS.
- N. Camaioni, G. Ridolfi, V. Fattori, L. Favaretto and G. Barbarella, Appl. Phys. Lett., 2004, 84, 1901 CrossRef CAS.
- G. Ridolfi, N. Camaioni, P. Samorì, M. Gazzano, G. Accorsi, N. Armaroli, L. Favaretto and G. Barbarella, J. Mater. Chem., 2005, 15, 895 RSC.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC.
- A. Beeby, K. Findlay, P. J. Low and T. B. Marder, J. Am. Chem. Soc., 2002, 124, 8280 CrossRef CAS.
- A. Beeby, K. Findlay, P. Low, T. B. Marder, P. Matousek, A. Parker, S. Rutter and M. Towrie, Chem. Commun., 2003, 2406 RSC.
- D. Lydon, L. Porrès, A. Beeby, T. B. Marder and P. Low, New J. Chem., 2005, 29, 972 RSC.
- S. Greaves, E. Flynn, E. Futcher, E. Wrede, D. Lydon, P. Low, S. Rutter and A. Beeby, J. Phys. Chem. A, 2006, 110, 2114 CrossRef CAS.
- A. Beeby, K. S. Findlay, A. E. Goeta, L. Porrès, S. R. Rutter and A. L. Thompson, Photochem. Photobiol. Sci., 2007, 6, 982 CAS.
- J. Siddle, R. Ward, J. Collings, S. Rutter, L. Porrès, L. Applegarth, A. Beeby, A. Batsanov, A. Thompson and J. Howard, New J. Chem., 2007, 31, 841 RSC.
- G. Zotti, G. Schiavon, S. Zecchin and A. Berlin, Synth. Met., 1998, 97, 245 CrossRef CAS.
- Y. Miyahara and T. Inazu, Tetrahedron Lett., 1990, 31, 5955 CrossRef CAS.
- R. Ballini, E. Marcantoni and M. Petrini, Tetrahedron Lett., 1992, 33, 4835 CrossRef CAS.
- E. Amir and S. Rozen, Angew. Chem., Int. Ed., 2005, 44, 7374 CrossRef CAS.
- N. Furukawa, H. Hoshiai, T. Shibutani, M. Higaki, F. Iwasaki and H. Fujihara, Heterocycles, 1992, 34, 1085 CrossRef CAS.
- F. Tran-Van, S. Garreau, G. Louarn, G. Froyer and C. Chevrot, J. Mater. Chem., 2001, 11, 1378 RSC.
- L. Fuller, B. Iddon and K. Smith, J. Chem. Soc., Perkin Trans. 1, 1997, 3465 RSC.
- A. Comel and G. Kirsch, J. Heterocycl. Chem., 2001, 38, 1167 CrossRef CAS.
- M. Gather, M. Heeney, W. Zhang, K. Whitehead, D. Bradley, I. McCulloch and A. Campbell, Chem. Commun., 2008, 1079 RSC.
- A. Köhler, J. Wilson, R. Friend, M. Al-Suti, M. Khan, A. Gerhard and H. Bässler, J. Chem. Phys., 2002, 116, 9457 CrossRef.
- P. Li, B. Ahrens, N. Feeder, P. Raithby, S. Teat and M. Khan, Dalton Trans., 2005, 874 RSC.
- N. Baek, S. Hau, H. Yip, O. Acton, K. Chen and A. Jen, Chem. Mater., 2008, 20, 5734 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schiegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Za- krzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, revision c.02., Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full synthetic procedures and characterisation of materials. CCDC reference numbers 839599–839604. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1ra00728a |
| ‡ It is with great sadness that we report the death of Dr Andrés Goeta on 29th July 2011. |
| This journal is © The Royal Society of Chemistry 2012 |