One-step hydrothermal synthesis of SnS2/graphene composites as anode material for highly efficient rechargeable lithium ion batteries†
Linhai
Zhuo
ac,
Yingqiang
Wu
acd,
Lingyan
Wang
acd,
Yancun
Yu
ac,
Xinbo
Zhang
*b and
Fengyu
Zhao
*ac
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China
bState Key Laboratory of Rare Earth Resource Utilizations, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, P. R. China. E-mail: xbzhang@ciac.jl.cn
cLaboratory of Green Chemistry and Process, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: zhaofy@ciac.jl.cn;; Fax: +86-431-85262410
dUniversity of the Chinese Academy of Sciences, Beijing, 100049, China
First published on 13th March 2012
Abstract
SnS2/graphene nanosheets (SnS2/GNS) composites were synthesized by a one-step hydrothermal method. The composites exhibit remarkably improved Li-storage ability with a good cycling life and high capability superior to that of the pure SnS2 counterpart due to a synergic effect between the graphene and SnS2 nanosheets.
Recently, various metal sulfides, including SnS2, MoS2, Cu2SnS3, CoS2, and SnS, have been studied as possible candidates for carbonaceous anode materials for lithium ion batteries (LIB) because of their higher capacities.1–3 Compared with SnO2 crystallized in a tetragonal structure, SnS2 has a layered hexagonal CdI2-type crystal structure which has swelling tolerant hosting spaces and can provide enhanced diffusion for lithium ions.1a,2b,4 This crystallographic feature is suitable for the intercalation of lithium ions and the compensation of the alloying/dealloying volume change.5 However, the capacity fading of SnS2 is serious, which is partly attributed to the problems of low electrical conductivity, dissolution of sulfur in the electrolyte and volume expansion during cycling.2e,4,6 To overcome these problems, many researchers are focusing on fabrication of SnS2 nanomaterials which are able to decrease their volume change and enhance the cycling life and rate capability. For example, various SnS2 nanostructures with different morphologies, such as nanosheets, nanoplates, nanorods, nanobelts, nanoflowers and nanotubes, have been prepared employing a variety of synthetic methods.2,5 Another way to improve the cycling stability of SnS2 is to synthesize nanostructured SnS2/carbon composites, which not only can buffer the internal volume change during cycling, but also can enhance the electronic conductivity of the composites.6,7
As a family member of carbon, graphene has drawn special attention due to its excellent electronic conductivity, mechanical properties and high surface area. Its two-dimensional (2D) nanostructure can match well with other materials crystallized in a layered structure, which provides opportunities for designing new functional composite materials through grafting the target materials on it. To date, zero-dimensional metals and metal oxides grafted on graphene have been prepared and showed promising electrochemical performances as anode materials for LIB.8,9 However, only a limited number of examples have been reported of the synthesis of 2D metal sulfides/graphene composites, especially for Sn-based sulfides.8,10,11 Recently, SnSe2 nanoplate/graphene composites were prepared through a two-step solution approach and applied as anode materials in LIB, exhibiting promising storage performance compared to that of SnSe2 nanoplates or graphene alone.8 Very recently, 2D graphene-SnS2 units were synthesized via a two-step approach by Luo et al. They obtained an enhanced rate capability compared to that of the other Sn-based/graphene composites as anodes in LIB reported in the literature.11
In this study, we employed a simple one-step hydrothermal route to synthesize SnS2/GNS composites. The synthesis was carried out by the thermal hydrolysis/dissociation of thioacetamide (TAA) with the presence of graphene oxide (GO) in aqueous solution of tin(IV) chloride pentahydrate at a suitable reaction temperature (see ESI†, Experimental section). As a result of many functional groups on the surface of GO, such as carboxyl, hydroxyl and epoxy groups, Sn4+ can tightly adsorb onto the GO surface via the electrostatic interaction.10 In the following process, the hydrolysis/dissociation reactions of TAA in the aqueous solution would generate sulfur ions and proceed faster at high temperature.5 The generated sulfur ions would bring the nucleation and promote the formation of SnS2/GNS composites, ultimately, during the hydrothermal period. Through the synthesis, the starting GO sheets were converted to GNS due to the hydrothermal treatment and the presence of sulfur ions released from TAA.10 For comparison, pristine SnS2 was also prepared in a similar process, but without GO.
The overall crystal structure and phase purity of the two as-obtained samples, pristine SnS2 and SnS2/GNS composites, were inspected by X-ray diffraction (XRD). As shown in Fig. 1a, the SnS2/GNS composites show a similar crystalline structure to that of pristine SnS2, and are in good agreement with those of standard XRD patterns of hexagonal SnS2 (JCPDS: 23-0677). Additionally, the intensity of the (001) plane at 2θ = 15.10 for SnS2/GNS composites is lower than that of pristine SnS2, indicating that the growth of the (001) plane for SnS2/GNS composites is inhibited due to the presence of graphene.10 The general morphology of SnS2/GNS composites is shown in Fig. 1b. It can be seen that the composites display a 2D nanosheet structure with thickness of about 20–25 nm. Without using GO in the synthetic process, the pristine SnS2 materials exhibit a characteristic of hexagonal platelet structure with discal size of 100 nm in thickness, which is much thicker than that of the SnS2/GNS composites (see ESI†, Fig. S1). The SnS2/GNS composites were also analysed by TEM. As observed from Fig. 1c, the 2D nanosheets of SnS2 match well with the surface of GNS, which displays a typical crinkly and rippled structure. The elemental mapping of C, S, and Sn by energy dispersive X-ray spectroscopy on SnS2/GNS also shows that SnS2 nanosheets are homogeneously distributed in the carbon matrix (see ESI†, Fig. S2). The carbon content is 9.95 wt% in the SnS2/GNS composites, analysed by an element analyzer. The crystal planes of the SnS2/GNS were also investigated by high-resolution TEM (HRTEM). Fig. 1d is the HRTEM image and the corresponding Fast Fourier Transform pattern (the inset image) of the top view of a nanosheet. The lattice fringes with a distance of 0.31 nm are clearly observed, which can be indexed as the (100) crystal plane of hexagonal SnS2.
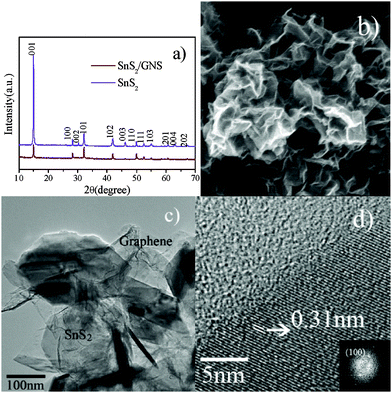 | ||
| Fig. 1 (a) XRD patterns of SnS2 and SnS2/GNS. (b) SEM image of SnS2/GNS nanostructures. (c) TEM image of SnS2/GNS nanostructures. (d) HRTEM image of SnS2/GNS nanostructures. | ||
The electrochemical performances of the pristine SnS2 nanoplates and SnS2/GNS composites were evaluated and compared. Cyclic voltammograms (CVs) of the pristine SnS2 and SnS2/GNS composites for two cycles are presented in Fig. 2a,b. The peaks at 1.1 and 1.5 V in the first cathodic sweep have been reported to correspond to the decomposition of the SnS2 nanoparticles into metallic tin and Li2S, while those at 0.1 V in the first cathodic scan and at ∼0.6 V in the first anodic scan are known to represent the redox peak couple of the reaction of Li ions subsequently reacted with Sn metal.4 The generated Li2S acts as an inert matrix surrounding the active Sn grains and could suppress the aggregation of the Li–Sn alloy.1a The oxidation peaks at ca. 1.8 V in the first anodic scan possibly originated from the oxygenation of Sn nanoparticles at higher potential in the charged state.3,10 The peaks at ca. 1.7 V in the first cathodic scan can be attributed to the lithium intercalation of the SnS2 layers without phase decomposition.4,7Fig. 2c shows the first and second charge–discharge curves of the SnS2/GNS composites at a current density of 100 mA g−1 and cycled between 3.0 and 0.01 V vs. Li/Li+. There are four plateaus observed at 2.0–1.7, 1.7–1.5, 1.5–1.3 and 0.5–0.01 V for the composites electrode in the first discharge, which is in accordance with the findings of pristine SnS2 (Fig. 2d). In the first charge (delithiation) process, two potential plateaus display at about 0.6 and 1.9 V. These results are also observed in the CVs of the composites (Fig. 2a). In addition, during the first charge/discharge process, the lithiation capacity of SnS2/GNS composites is 1367 mAh g−1, and the delithiation capacity reaches 1145 mAh g−1, which is much higher than the capacity of free SnS2.
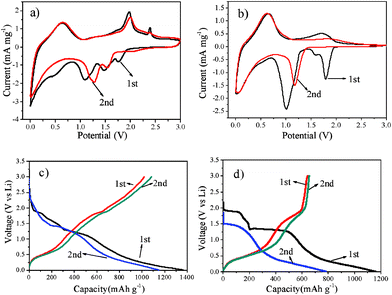 | ||
| Fig. 2 Cyclic voltammograms (CVs) of SnS2/GNS electrodes (a) and SnS2 electrodes (b) at a scan rate of 0.5 mV s−1 during the first two cycles. The first two charge and discharge curves of SnS2/GNS (c) and SnS2 (d) at a current density of 100 mA g−1. | ||
Fig. 3a shows the typical results of cycling of SnS2/GNS composites and pristine SnS2 nanoplate electrodes cycled between 3 V and 0.01 V with cycling performances up to 30 cycles at a rate of 100 mA g−1. The SnS2/GNS composites have a large reversible capacity of 1158 mAh g−1, while still retaining a high reversible capacity of 1114 mAh g−1 after 30 cycles. The coulombic efficiency increases from 83% in the first cycle to over 97% in the subsequent cycles. Whereas the pristine SnS2 nanoplates show poor cycle performance with an initial reversible capacity of 783 mAh g−1, followed by a gradual reduction to 506 mAh g−1 after 30 cycles. The high reversible capacity and excellent cycling performance of the SnS2/GNS composites are also exhibited in the rate capability. The SnS2/GNS composites and pristine SnS2 nanoplates were tested with a variable C rate (from 100 mA g−1 to 1000 mA g−1). Fig. 3b shows the rate cycling behavior of SnS2/GNS and pristine SnS2 nanoplate electrodes. The SnS2/GNS composites also present good rate performance. Even at a high current density of 1000 mA g−1, the specific capacity remains at ca. 870 mAh g−1, which is still much higher than that of free SnS2. Additionally, the extraordinary cycling stability of the composites is exhibited at various current densities. Even though the current changes from 1000 mA g−1 to 100 mA g−1, the specific capacity of the composites reverts to ca. 1100 mA g−1 and does not visibly change in the following cycles. Compared with the composites, the pristine SnS2 nanoplates also manifest better rate capability in spite of their relative lower capacity. This should be attributed to the nanoscale lateral size of the platelets and the barrier & buffer role of generated Li2S during cycling.
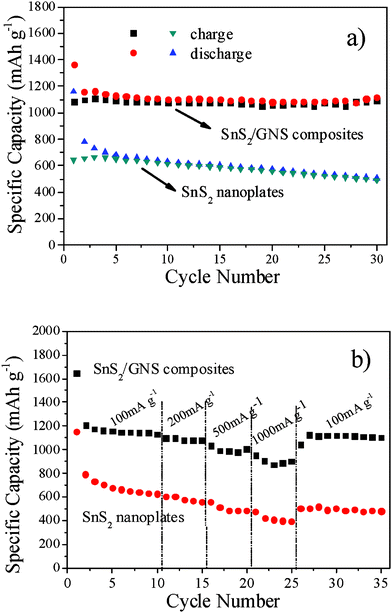 | ||
| Fig. 3 (a) Cycling behaviour of SnS2 and SnS2/GNS electrodes at a current density of 100 mA g−1. (b) Discharge capacity vs. cycle number of SnS2/GNS and SnS2 nanoplate electrodes at various current densities. | ||
In order to further study the improved high-rate performance, electrochemical impedance spectroscopy measurements on SnS2/GNS composites and pristine SnS2 nanoplate electrodes were performed and compared. Fig. 4 shows the impedance plots of the SnS2/GNS composites and SnS2 nanoplates with a characteristic of semicircles in the high frequency region and straight sloping lines in the low frequency region. From the Nyquist plots, the charge transfer resistance (Rct) of the SnS2/GNS electrode (27 Ω), closely related to the reaction in the electrode/electrolyte interface, is less than that of pristine SnS2 electrode (81 Ω). This may originate from the fast transfer rate of Li+ resulting from the improved surface electronic conductivity of SnS2/GNS composites.4
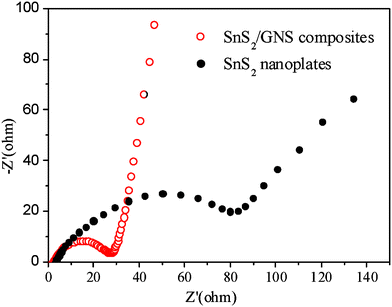 | ||
| Fig. 4 Electrochemical impedance spectra of SnS2/GNS composites and pristine SnS2 nanoelectrodes. | ||
It should be noted that not only SnS2 but also GNS contribute to the capacity. The pristine SnS2 nanoplates have an initial reversible capacity of 783 mAh g−1, while the bare GNS show an initial discharge capacity of 1347 mAh g−1 and charge capacity of 744 mAh g−1 with a coulombic efficiency of 55% (Fig. S3†). However, when only a small fraction of graphene was added to synthesize the SnS2/GNS composites, the reversible capacity was enhanced to 1158 mAh g−1 and the rate capability was increased to 870 mAh g−1 at a rate of 1000 mA g−1. This may be attributed to a synergic effect between the graphene and SnS2 nanosheets.10,11 Firstly, GNS have intrinsic properties such as high surface area and excellent flexibility, which not only can supply enough void space to absorb volume change, but also can prevent the aggregation of SnS2 particles during the process of delithiation and lithiation. Secondly, as electronic conductive channels in hybrid material, GNS can improve the electronic conductivity of the composites. Thirdly, the thickness of the SnS2 is confined owing to the presence of graphene, which facilitates faster lithium ion diffusion through the active materials.
In summary, SnS2/GNS composites were synthesized by a simple one-step hydrothermal method. The reversible capacities and cycling performance are significantly improved in comparison with those of the pristine SnS2. After 30 cycles, the reversible capacity of SnS2/GNS could still be retained at 1114 mAh g−1 with the retention of about 96%. The high capacity, cycling stability and rate capability of the SnS2/GNS composites can be attributed to the intimate interaction between the graphene substrates and SnS2 layers grafted on them.
Acknowledgements
This work was financially supported by NSFC20873139, KJCX2, YWH16, and 20086063.References
- (a) Y. Li, J. Tu, X. Huang, H. Wu and Y. Yuan, Electrochim. Acta, 2006, 52, 1383 CrossRef CAS; (b) J. Z. Wang, S. L. Chou, S. Y. Chew, J. Z. Sun, M. Forsyth, D. R. MacFarlane and H. K. Liu, Solid State Ionics, 2008, 179, 2379 CrossRef CAS; (c) G. D. Du, Z. P. Guo, S. Q. Wang, R. Zeng, Z. X. Chen and H. K. Liu, Chem. Commun., 2010, 46, 1106 RSC; (d) Q. H. Wang, L. F. Jiao, Y. Han, H. M. Du, W. X. Peng, Q. N. Huan, D. W. Song, Y. C. Si, Y. J. Wang and H. T. Yuan, J. Phys. Chem. C, 2011, 115, 8300 CrossRef CAS.
- (a) J. Seo, J. Jang, S. Park, C. Kim, B. Park and J. Cheon, Adv. Mater., 2008, 20, 4269 CrossRef CAS; (b) K. Chang, W. X. Chen and H. Li, Electrochim. Acta, 2011, 56, 2856 CrossRef CAS; (c) A. Yella, E. Mugnaioli, M. Panthofer, H. A. Therese, U. Kolb and W. Tremel, Angew. Chem., Int. Ed., 2009, 48, 6426 CrossRef CAS; (d) Y. Lei, S. Song, W. Fan, Y. Xing and H. Zhang, J. Phys. Chem. C, 2009, 113, 1280 CrossRef CAS; (e) T. Takeuchi, H. Sakaebe, H. Kageyama, K. Handa, T. Sakai and K. Tatsumi, J. Electrochem. Soc., 2009, 156, A958 CrossRef CAS.
- B. Qu, M. Zhang, D. Lei, Y. Zeng, Y. Chen, L. Chen, Q. Li, Y. Wang and T. Wang, Nanoscale, 2011, 3, 3646 RSC.
- (a) T. J. Kim, C. Kirn, D. Son, M. Choi and B. Park, J. Power Sources, 2007, 167, 529 CrossRef CAS; (b) S. Liu, X. Yin, L. Chen, Q. Li and T. Wang, Solid State Sci., 2010, 12, 712 CrossRef CAS; (c) J. T. Zai, K. X. Wang, Y. Z. Su, X. F. Qian and J. S. Chen, J. Power Sources, 2011, 196, 3650 CrossRef CAS; (d) Y. Zhao, J. Li, Y. Ding and L. Guan, RSC Adv., 2011, 1, 852 RSC.
- C. X. Zhai, N. Du, H. Zhang and D. R. Yang, Chem. Commun., 2011, 47, 1270 RSC.
- Y. Li, J. P. Tu, X. H. Huang, H. M. Wu and Y. F. Yuan, Electrochem. Commun., 2007, 9, 49 CrossRef CAS.
- (a) H. S. Kim, Y. H. Chung, S. H. Kang and Y. E. Sung, Electrochim. Acta, 2009, 54, 3606 CrossRef CAS; (b) M. Noh, Y. Kwon, H. Lee, J. Cho, Y. Kim and M. G. Kim, Chem. Mater., 2005, 17, 1926 CrossRef CAS.
- J. Choi, J. Jin, I. G. Jung, J. M. Kim, H. J. Kim and S. U. Son, Chem. Commun., 2011, 47, 5241 RSC.
- J. Zhou, H. Song, L. Ma and X. Chen, RSC Adv., 2011, 1, 782 RSC.
- (a) K. Chang and W. Chen, Chem. Commun., 2011, 47, 4252 RSC; (b) K. Chang and W. X. Chen, ACS Nano, 2011, 5, 4720 CrossRef CAS; (c) K. Chang and W. X. Chen, J. Mater. Chem., 2011, 21, 17175 RSC.
- B. Luo, Y. Fang, B. Wang, J. Zhou, H. Song and L. Zhi, Energy Environ. Sci., 2012, 5, 5226 CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra00002d |
| This journal is © The Royal Society of Chemistry 2012 |