Monosubstituted dually cationic cyclodextrins for stronger chiral recognition†
Jie
Zhou‡
,
Yun
Dai‡
,
Shuye
Wang
,
Enwei
Zhu
,
Jiefeng
Hai
,
Yun
Liu
,
Jian
Tang
* and
Weihua
Tang
*
Key Laboratory of Soft Chemistry and Functional Materials, Ministry of Education, Nanjing University of Science and Technology, Nanjing, P. R. China.. E-mail: tangj2005@hotmail.com; whtang@mail.njust.edu.cn; Fax: +86 25 8431 7311; Tel: +86 25 8431 7311
First published on 13th March 2012
Abstract
Novel monosubstituted dually cationic cyclodextrins (CDs) have been synthesized by anchoring different alkyl chain spaced imidazolium and ammonium sidearm onto the CD primary ring. These cationic CDs exhibit satisfactory enantioselectivities for amino acids and acidic racemates in aqueous capillary electrophoresis.
Cyclodextrins (CDs) are a family of macrocyclic oligosaccharides composed of six to twelve D-glucopyranose units.1,2 Featuring a hydrophilic outer surface and a hydrophobic inner cavity, CDs have been widely used in molecular recognition due to their capability to form inclusion complexes with a variety of aromatic molecules of suitable size.3,4 Among the CD derivatives developed so far, cationic CDs have attracted considerable interest in separation science,5–10 sorption,11 and drug/gene delivery.12
With cationic CDs, improved enantioselectivities have been achieved for negatively charged analytes due to the synergistic effects of electrostatic interactions and inclusion complexation for chiral recognition.2,5,6 For repeatability and reproducibility in synthesis and application, a strategy of using structurally defined CDs is recommended.5–10 Many amine/ammonium-substituted CDs have demonstrated good enantioselectivities for amino acids like ampholytic racemates, but were limited for acidic racemates, while imidazole-substituted CDs performed in an exactly opposite manner.8–10 Triethylammonium CD based sorption systems exhibited strong affinity toward aromatic compounds via inclusion complex formation in the hydrophobic cavities.11 By mixing an aqueous solution of anionic copolymers of glucuronic acid and starch with a cationic thioether of a CD, stable spherical nanoparticles were formulated to conjugate with pteroic acid as a cell-specific ligand for targeting cancer cells.12a A great number of strategies to promote interactions between cationic CD conjugates with genetic material have been employed by fully exploiting the inside–outside/upper–lower face anisotropy of the CD nanometric platform. Covalent modification, self-assembling and supramolecular ligation are put forward with the ultimate goal to build artificial viruses for programmed and efficient gene therapy.12b
Aiming to design versatile cationic CDs for improved molecular recognition for both ampholytic and acidic racemates, we report herein the preparation of monosubstituted dually cationic CD to incorporate both imidazolium and ammonium moieties in the sidearm on the CD's primary ring. In our current synthesis, an alkylamine linked imidazole moiety is directly introduced onto the C6 of the glucopyranose subunit, which offers dually cationic CDs with high aqueous solubility and extra electrostatic interactions besides inclusion complexation for molecular recognition.
The synthetic route for dually cationic CDs is shown in Scheme 1. Using the general Gabriel synthesis,13 (bromoalkyl)phalimides 3 was reacted with imidazole in the presence of NaH to give N-[(1H-imidazolyl)alkyl]phthalimide 4. A key step of nucleophilic attack of p-tolylsulfonyl β-cyclodextrin 1 by derivative 49,10 afforded the key monosubstituted CD intermediate 5. The phthalimide removal with hydrazine monohydrate gave mono-6A-[3-(aminoalkyl)-imidazol-1-ium]-6A-deoxy-β-CD tosylate 6 with a yield of 60–81%. Further anion exchange of 6 with Amberlie (Cl) resin and treatment with dilute hydrochloric acid gave the final product, mono-6A-[3-(ammonioalkyl)-imidazol-1-ium]-6A-deoxy-β-CD chloride 7 as white solid. Featuring two cationic centers in their monosubstituted sidearms, these CDs 7 are endowed with high aqueous solubility and permanent charge with the immidazolium moiety regardless of solution pH, which is advantageous to tune the chiral recognition window for oppositely charged guest molecules.
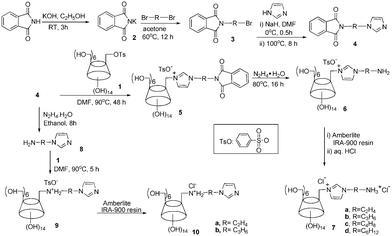 | ||
| Scheme 1 Synthetic route for dually cationic CDs. | ||
By deprotection of N-[(1H-imidazolyl)alkyl]phthalimide 4 with hydrazine,13d (1H-imidazol-1-yl)alkylamine 8 was first obtained, which can undergo nucleophilic attack of p-tolylsulfonyl β-cyclodextrin 1 to afford ammonium-substituted cationic CD 9. Similar anionic exchange offered the second series of dually cationic CDs in good yields.
With the dually cationic CDs at hand, their potential in chiral separation was explored by taking mono-6A-[3-(4-ammoniobutyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin chloride (7c, AMBIMCD) as a model chiral selector for chiral CE. As reported in the literature, it is very challenging for single cationic CDs to achieve versatile enantioselectivities for both ampholytic and acidic racemates. In this study, eight dansylated amino acids and seven acidic compounds (structures in Fig. 1) were selected as model analytes. The enantioseparation experiments were run in pH 6.0 phosphate buffer to ensure the full deprotonation of the analytes.9,10 The migration time of the first enantiomer, selectivity (α) and chiral resolution (Rs) values for all analytes achieved with 1.0 mM and 2.5 mM AMBIMCD are summarized in Table 1.
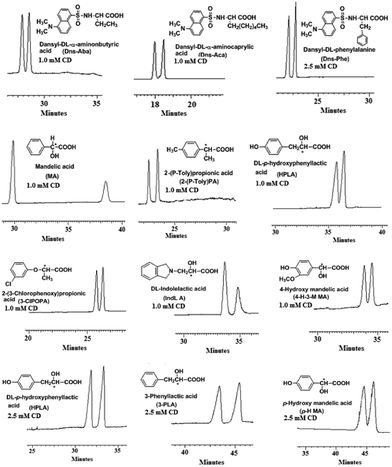 | ||
| Fig. 1 Typical electropherograms with either 1.0 mM or 2.5 mM AMBIMCD at pH 6.0. | ||
| Analytes | 1.0 mM | 2.5 mM | ||||
|---|---|---|---|---|---|---|
| t 1 | α | R s | t 1 | α | R s | |
| a α = (t2 − tEOF)/(t1 − tEOF), 2,4-dichloro PPA: 2-(2,4-dichlorophenoxy)propionic acid. | ||||||
| Dns–Aba | 23.5 | 1.04 | 1.1 | 27.9 | 1.05 | 1.0 |
| Dns–Aca | 18.0 | 1.09 | 1.8 | 25.0 | 1.12 | 1.7 |
| Dns–DL-methionine | 21.2 | 1.02 | 0.9 | 28.8 | 1.03 | 2.1 |
| Dns–DL-norleucine | 18.3 | 1.02 | 0.8 | 26.1 | 1.04 | 0.9 |
| Dns–DL-norvaline | 26.6 | 1.03 | 1.4 | 26.4 | 1.04 | 2.6 |
| Dns–Phe | 17.8 | 1.05 | 0.7 | 22.0 | 1.08 | 1.5 |
| Dns–DL-serine | 20.8 | 1.02 | 0.6 | 28.5 | 1.03 | 0.7 |
| Dns–DL-threonine | 20.1 | 1.04 | 1.3 | 29.4 | 1.07 | 2.7 |
| 2,4-Dichloro PPA | 21.7 | 1.08 | 2.0 | 45.6 | 1.09 | 3.6 |
| MA | 29.9 | 1.43 | 8.1 | 58.2 | 1.12 | 12.2 |
| 2-(p-Tolyl) PA | 22.3 | 1.05 | 1.6 | 31.6 | 1.06 | 3.2 |
| 4-H-3-M MA | 33.8 | 1.03 | 1.1 | 42.1 | 1.10 | 1.6 |
| p-H MA | 22.1 | 1.03 | 2.8 | 44.2 | 1.05 | 3.5 |
| p-HPLA | 35.8 | 1.05 | 1.8 | 31.8 | 1.04 | 1.5 |
| 3-PLA | 30.6 | 1.05 | 1.2 | 43.4 | 1.06 | 1.8 |
As shown, AMBIMCD exhibits versatile good enantioselectivities for ampholytic acid and acidic compounds, with 13 racemate baselines separated (Rs > 1.0) at 2.5 mM CD, in comparison to most cationic CDs reported so far.6,14–16 The chiral resolution of racemates improved at higher CD concentration. AMBIMCD presented better enantioselectivities for Dns–DL-norvaline, Dns–Phe, Dns–DL-threonine than CD-mh.11 The Rs of 2.6 for Dns–DL-norvaline with 2.5 mM AMBIMCD is even higher than those achieved by all our reported imidazolium CDs at the concentration of 3 mM.17 Compared with the dicationic CDs15 reported by Marchelli and coworkers, our AMBIMCD exhibited better resolution towards some Dns–amino acids and MA at similar reparation conditions. In addition, the Rs values of Dns–Aba, Dns–Aca, Dns–Phe and Dns–Thr achieved with 2.5 mM AMBIMCD are higher than those with 5.0 mM of our reported butylammonium CD.9c Impressively, AMBIMCD exhibits good to excellent enantioselectivities for acidic racemates, even at 1.0 mM concentration, with the Rs value of mandelic acid hitting 8.1. As known to all, improved chiral recognition of charged CD molecules for oppositely charged racemates is based on the formation of both inclusion complexes and electrostatic interactions. Our designed CDs boast two cationic centers, which may enhance their enenatioselectivities due to stronger driving forces, which are reflected by the good enantioselectivities of AMBuIMCD for both Dns–amino acids and acidic racemates. Representative electropherograms of chiral separation with 1.0 or 2.5 mM AMBIMCD are plotted in Fig. 1.
A close look at the migration time of analytes shows that enantioseparation with AMBuIMCD takes a slightly longer time than our previously reported sing cationic CDs,9,10 which may be explained by the more cationic nature of the selector/analyte complexes in this case. Hence, the formation degree is lower to compensate for their more positive charge.
The inclusion complexation behavior was investigated by ESI-MS and mono-dimensional 1H NMR measurements (TOCSY).15 With the soft ionization technique, ESI-MS, it is possible to transfer existing ions to the gaseous phase without breaking nonconvalent interactions.18 It is thus very useful for the determination of the formation and stoichiometry of our cationic CD inclusion complexes. By taking mono-6A-[3-(6-ammoniohexyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin chloride (7d, AMHIMCD) as the CD host and single enantiomers of 3-phenyllactic acid (3-PLA) (i.e. R-3-PLA and S-3-PLA) as mode guests, the inclusion complexation mode of AMHIMCD/R-3-PLA and AMHIMCD/S-3-PLA complexes (10 mM) was revealed with 1H NMR measurement in D2O (pD 6.0). Analysis of an aqueous solution of AMHIMCD and 3-PLA by ESI-MS revealed the formation of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex between the deprotonated 3-PLA and AMHIMCD cation at m/z 1450. As shown in Fig. 2, when R-3-PLA was mixed with CD-X in aqueous solution, the split of the homologs of benzene were changed. The overlapped Hp and Hm proton signals for pure R-3-PLA were well separated to present extended splitting peaks in AMHIMCD/R-3-PLA. Clear doublet splitting is observed for both Hm and Hp, while a doublet doublet splitting was observed for Ho due to the coupling effects from both Hm and Hp.
:1 complex between the deprotonated 3-PLA and AMHIMCD cation at m/z 1450. As shown in Fig. 2, when R-3-PLA was mixed with CD-X in aqueous solution, the split of the homologs of benzene were changed. The overlapped Hp and Hm proton signals for pure R-3-PLA were well separated to present extended splitting peaks in AMHIMCD/R-3-PLA. Clear doublet splitting is observed for both Hm and Hp, while a doublet doublet splitting was observed for Ho due to the coupling effects from both Hm and Hp.
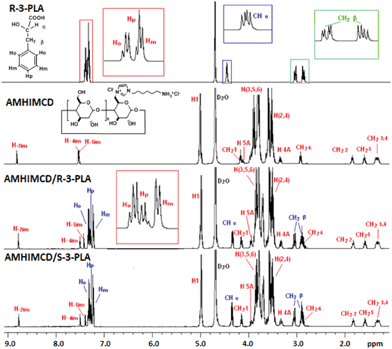 | ||
| Fig. 2
1H NMR spectra of AMHIMCD, R-3-PLA and the mixture of AMHIMCD/R-3-PLA and AMHIMCD/S-3-PLA (1:1, 10 mM, pD 6.0). | ||
With a close look at the chemical shift for the protons of guest molecules in their complexes with AMHIMCD, the proof for molecular recognition can be found. High-field shifted chemical shifts obviously observed for –CHα and CH2β of R-3-PLA are mainly attributed to the electrostatic interactions formed between R-3-PLA and the CD. Down-field shifted chemical shifts for the aromatic moiety of 3-PLA are also visible, mainly due to the shield effect of the CD cavity. Among the three kinds of aromatic protons, Ho exhibited the largest chemical shift increment. All these demonstrate that the benzene ring and CD formed the inclusion complexes. This recognition behavior of our dually cationic CDs may find potential applications in drug delivery and asymmetric template synthesis.2,19
In summary, we have developed dually cationic CDs, with Gabriel synthesis as a key step, which exhibit good enantioselectivities for both ampholytic and acidic racemates in aqueous CE. A chiral resolution of 8.1 was achieved for mandelic acid, with only 1.0 mM AMBIMCD. This synthetic methodology may be readily applicable to design new host molecules by constructing stronger driving forces for molecular recognition.
Experimental section
All reagents were of analytical grade quality as obtained from commercial suppliers and used without further purification. Ethanol and DMF were freshly dried prior to use using standard procedures. Compounds 1 and 2 were synthesized according to reported procedures.2,13 Detailed synthesis of 3, 4, 8a and 10a is described in the ESI.†Mono-6A-[3-(2-(1,3-dioxoisoindolin-2-yl)ethyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (5a)
To a solution of TsCD 1 (5.61 g, 4.36 mmol) in DMF (10 mL) was added 4a (3.15 g, 13.07 mmol) under vigorous stirring. The reaction mixture was refluxed at 90 °C for 48 h under N2. After cooling down the reaction mixture to room temperature, the crude product was precipitated out from acetone (40 mL). A further recrystallization from water afforded the title compound 5a (4.47 g, 67%) as a white solid: m.p. ∼225.2 °C; IR (cm−1, KBr): 3391(O–H str), 2928 (C–H str), 1717 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str), 1637 and 1624 (CC str), 1157 (SO str), 1080 and 1032 (C–O–C str); 1H NMR (500 MHz, DMSO-d6): δ = 9.15 (s, 1H, CH-2im), 7.89 (s, 1H, CH-4im), 7.86 (m, 2H, CH-aromatic), 7.68 (s, 1H, CH-5im), 7.47(d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.11 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.65–5.81 (m, 14H, OH-2CD and OH-3CD), 4.78–4.92 (m,7H, H-1CD), 4.43–4.53 (m, 5H, OH-6CD), 4.02 (m, 1H, H-3′CD), 3.86 (m, 1H, H-5′CD), 3.76 (m, 2H, –CH2-N-im), 3.42–3.64 (m, 26H, H-5CD, H-3CD and H-6CD), 3.18–3.37 (m, 14H, H-2CD and H-4CD), 3.01–3.03 (m, 2H, –CH2-N-imine), 2.28 (s, 3H, –CH3-OTs); 13C NMR (125 MHz, D2O): δ = 164.93, 142.14, 135.85, 135.08, 130.93, 130.33, 129.05, 125.36, 124.29, 123.44, 122.07, 102.27, 101.03, 81.30, 73.50, 72.58, 71.39, 60.16, 37.64, 31.41, 20.53; ESI-MS (m/z): calculated for [M+] 1359.22, found 1358.67; Anal. Calcd for C62H87O39N3S: C 48.66, H 5.73, N 2.75; found: C 48.78, H 5.72, N 2.62.
O str), 1637 and 1624 (CC str), 1157 (SO str), 1080 and 1032 (C–O–C str); 1H NMR (500 MHz, DMSO-d6): δ = 9.15 (s, 1H, CH-2im), 7.89 (s, 1H, CH-4im), 7.86 (m, 2H, CH-aromatic), 7.68 (s, 1H, CH-5im), 7.47(d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.11 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.65–5.81 (m, 14H, OH-2CD and OH-3CD), 4.78–4.92 (m,7H, H-1CD), 4.43–4.53 (m, 5H, OH-6CD), 4.02 (m, 1H, H-3′CD), 3.86 (m, 1H, H-5′CD), 3.76 (m, 2H, –CH2-N-im), 3.42–3.64 (m, 26H, H-5CD, H-3CD and H-6CD), 3.18–3.37 (m, 14H, H-2CD and H-4CD), 3.01–3.03 (m, 2H, –CH2-N-imine), 2.28 (s, 3H, –CH3-OTs); 13C NMR (125 MHz, D2O): δ = 164.93, 142.14, 135.85, 135.08, 130.93, 130.33, 129.05, 125.36, 124.29, 123.44, 122.07, 102.27, 101.03, 81.30, 73.50, 72.58, 71.39, 60.16, 37.64, 31.41, 20.53; ESI-MS (m/z): calculated for [M+] 1359.22, found 1358.67; Anal. Calcd for C62H87O39N3S: C 48.66, H 5.73, N 2.75; found: C 48.78, H 5.72, N 2.62.
Mono-6A-[3-(3-(1,3-dioxoisoindolin-2-yl)propyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (5b)
White solid (yield 63%): m.p. ∼234.3 °C; 1H NMR (500 MHz, DMSO-d6): δ = 8.93 (s, 1H,CH-2im), 8.17 (s, 1H, CH-4im), 7.82 (m, 2H, CH-aromatic), 7.67 (s, 1H, CH-5im), 7.48 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.13 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.64–5.79 (m, 14H, OH-2CD and OH-3CD), 4.88 (s, 1H, H-1′CD), 4.83 (s, 6H, H-1CD), 4.46–4.53 (m, 6H, OH-6CD), 4.16 (m, 2H, –CH2-N-im), 3.92 (m, 1H, H-3′CD), 3.82 (m, 1H, H-5′CD), 3.54–3.65 (m, 26H, H-5CD, H-3CD and H-6CD), 3.17–3.54 (m, 14H, H-2CD and H-4CD), 3.03 (m, 2H, –CH2-N-imine), 2.26 (s, 3H, –CH3-OTs), 1.65 (m, 2H, –CH2); 13C NMR (125 MHz, D2O): δ = 169.70, 141.48, 141.08, 135.79, 135.08, 131.12, 128.93, 125.40, 123.85, 123.37, 122.71, 120.99, 102.03, 81.93, 73.21, 72.66, 71.59, 60.14, 46.08, 35.39, 31.41, 27.71, 20.48; ESI-MS (m/z): calculated for [M+] 1373.25, found 1372.58; Anal. Calcd for C63H89O39N3S: C 48.99, H 5.81, N 2.72; found: C 49.02, H 5.83, N 2.66.
Mono-6A-[3-(4-(1,3-dioxoisoindolin-2-yl)butyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (5c)
White solid (yield 78%): m.p. ∼238.7 °C; 1H NMR (500 MHz, DMSO-d6): δ = 8.85 (s, 1H,CH-2im), 8.22 (s, 1H, CH-4im), 7.83 (m, 2H, CH-aromatic), 7.66 (s, 1H, CH-5im), 7.46 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.10 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.64–5.80 (m, 14H, OH-2CD and OH-3CD), 4.78–4.92 (m, 7H, H-1CD), 4.43–4.53 (m, 6H, OH-6CD), 4.18 (m, 1H, H-3′CD), 3.92 (m, 1H, H-5′CD), 3.86 (m, 2H, –CH2-N-im), 3.54–3.65 (m, 26H, H-5CD, H-3CD and H-6CD), 3.32–3.41 (m, 14H, H-2CD and H-4CD), 3.17 (m, 2H, –CH2-N-imine), 2.29 (s, 3H, –CH3-OTs) 1.83(m, 2H, –CH2), 1.53 (m, 2H, –CH2); 13C NMR (125 MHz, D2O): δ = 164.92, 144.14, 137.31, 133.47, 133.09, 130.38, 128.82, 124.53, 123.87, 122.71, 120.37, 102.03, 81.89, 73.27, 72.48, 70.59, 61.01, 47.99, 36.39, 31.71, 24.72, 21.06; ESI-MS (m/z): calculated for [M+] 1386.48, found 1386.71; Anal. Calcd for C64H91O39N3S: C 49.32, H 5.89, N 2.70; found: C 49.52, H 5.94, N 2.62.
Mono-6A-[3-(6-(1,3-dioxoisoindolin-2-yl)hexyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (5d)
White solid (yield 63%): m.p. ∼253.7 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.08 (s, 1H,CH-2im), 7.86 (m, 2H, CH-aromatic), 7.78 (s, 1H, CH-4im), 7.72 (s, 1H, CH-5im), 7.48(d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.12 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.64–5.84 (m, 14H, OH-2CD and OH-3CD), 4.81–4.85 (m,7H, H-1CD), 4.43–4.56 (m, 6H, OH-6CD), 4.14 (m, 2H, –CH2-N-im), 3.97 (m, 1H, H-3′CD), 3.83 (m, 1H, H-5′CD), 3.54-3.65 (m, 26H, H-5CD, H-3CD and H-6CD), 3.28–3.44 (m, 14H, H-2CD and H-4CD), 3.21 (m, 1H,H-4′CD), 3.05 (m, 2H, –CH2-N-imine), 1.81 (m, 2H, –CH2), 1.58 (m, 2H, –CH2), 1.29 (m, 4H, –CH2); 13C NMR (125 MHz, D2O): δ = 164.03, 160.78, 156.64, 141.68, 132.63, 129.10, 128.94, 125.79, 125.24, 123.57, 122.52; ESI-MS (m/z): calculated for [M+] 1415.33, found 1415.20; Anal. Calcd for C66H95O39N3S: C 49.97, H 6.04, N 2.65; found: C 50.02; H 6.02; N 2.60.
Mono-6A-[3-(2-aminoethyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (6a)13
A mixture solution of 5a (2.40 g, 1.57 mmol), ethanol (25 mL), and hydrazine hydrate (0.157 g, 3.14 mmol) was stirred at 80 °C for 16 h. Cooling down the reaction mixture to room temperature, the white insoluble stuff was removed by filtration and washed with cold ethanol (3 × 10 mL). The collected filtrate was concentrated to ∼5 mL. Precipitated out of dichloromethane, the title compound, mono-6A-[3-(2-aminoethyl)-imidazolium]-6A-deoxy-β-cyclodextrin tosylate 6a (1.78 g, 81%) was afforded as a white solid: d.p. (decomposition temperature) ∼239.4 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.04 (s, 1H,CH-2im), 7.75 (s, 1H, CH-4im), 7.71 (s, 1H, CH-5im), 7.46 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.11 (d, 2H, J = 9.0 Hz, CH-meta-OTs ), 5.66–5.79 (m, 14H, OH-2CD and OH-3CD), 4.77–4.86 (m,7H, H-1CD), 4.43–4.56 (m, 6H, OH-6CD), 4.13 (m, 1H, H-3′CD), 3.99 (m, 1H, H-5′CD), 3.84 (m, 2H, –CH2-N-im), 3.51-3.64 (m, 26H, H-5CD, H-3CD and H-6CD), 3.28–3.45 (m, 14H, H-2CD and H-4CD), 3.02 (m, 2H, –CH2-N-amino), 2.28(s, 3H, –CH3-OTs); 13C NMR (125 MHz, D2O): δ = 142.69, 135.02,129.21, 125.42, 123.37, 121.31, 119.26, 101.93, 82.09, 81.11, 73.21, 72.12, 71.12, 60.25, 44.47, 37.09, 20.58; ESI-MS (m/z): calculated for [M+] 1228.45, found 1228.53; Anal. Calcd for C54H85O37N3S: C 46.32, H 6.12, N 3.00; found: C 46.41, H 6.15, N 2.98.
Mono-6A-[3-(3-aminopropyl)-imidazolium]-6A-deoxy-β-cyclodextrin tosylate (6b)
White solid (yield 76%): d.p. ∼226.1 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.04 (s, 1H,CH-2im), 8.06 (s, 1H, CH-4im), 7.81 (s, 1H, CH-5im), 7.48 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.12 (d, 2H, J = 9.0 Hz, CH-meta-OTs ), 5.66–5.81 (m, 14H, OH-2CD and OH-3CD), 4.81–4.85 (m, 7H, H-1CD), 4.36–4.52 (m, 6H, OH-6CD), 3.88 (m, 1H, H-5′CD), 3.81 (m, 2H, –CH2-N-im), 3.55–3.67 (m, 26H, H-5CD, H-3CD and H-6CD), 3.35–3.46 (m, 14H, H-2CD and H-4CD), 3.14 (m, 2H, –CH2-N-amino), 2.28(s, 3H, –CH3-OTs), 1.93 (m, 2H, –CH2); 13C NMR (125 MHz, D2O): δ = 146.19, 137.23, 130.31, 126.22, 123.58, 122.27, 119.13, 101.8, 82.09, 81.15, 73.07, 72.04, 71.81, 60.27, 44.32, 30.81, 27.37, 21.06; ESI-MS (m/z): calculated for [M+] 1242.46, found 1242.56; Anal. Calcd for C55H87O37N3S: C 46.71, H 6.20, N 2.97; found: C 46.74; H 6.23, N 2.94.
Mono-6A-[3-(4-aminobutyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (6c)
White solid (yield 60%): d.p. ∼216.1 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.12 (s, 1H,CH-2im), 7.84 (s, 1H, CH-4im), 7.76 (s, 1H, CH-5im), 7.47 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.12 (d, 2H, J = 9.0 Hz, CH-meta-OTs ), 5.60–5.81 (m, 14H, OH-2CD and OH-3CD), 4.83–4.89 (m,7H, H-1CD), 4.42–4.50 (m, 5H, OH-6CD), 4.01 (m, 2H, –CH2-N-im), 3.85 (m, 1H, H-5′CD), 3.50–3.75 (m, 27H, H-5CD, H-3CD and H-6CD), 3.28–3.43 (m, 13H, H-2CD and H-4CD), 3.19 (m, 1H, H-4CD'), 3.04 (m, 2H, –CH2-N-amino), 2.29 (s, 3H, –CH3-OTs), 1.87 (m, 2H, –CH2), 1.51 (m, 2H, –CH2); 13C NMR (125 MHz, D2O): δ = 142.69, 135.02, 129.21, 125.42, 123.38, 121.32, 119.26, 101.93, 82.09, 81.30, 73.21, 72.13, 71.12, 60.23, 44.47, 37.09, 30.32, 24.78, 20.58; ESI-MS (m/z): calculated for [M+] 1256.48, found 1256.61; Anal. Calcd for C56H89O37N3S: C 47.09, H 6.28, N 2.94; found: C 47.89; H 6.32; N 2.91.
Mono-6A-[3-(6-aminohexyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin tosylate (6d)
White solid (yield 60%): d.p. ∼208 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.12 (s, 1H,CH-2im), 7.79 (s, 1H, CH-4im), 7.74 (s, 1H, CH-5im), 7.47 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 7.11 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.73–5.93 (m, 14H, OH-2CD and OH-3CD), 4.78–4.91 (m,7H, H-1CD), 4.36–4.51 (m, 6H, OH-6CD), 4.14 (m, 2H, –CH2-N-im), 3.98 (m, 1H, H-3′CD), 3.84 (m, 1H, H-5′CD), 3.55–3.66 (m, 26H, H-5CD, H-3CD and H-6CD), 3.29–3.37 (m, 13H, H-2CD and H-4CD), 3.21 (m, 1H, H-4CD'), 2.56 (m, 2H, –CH2-N-amino), 2.29 (s, 3H, –CH3-OTs), 1.81 (m, 2H, –CH2), 1.36 (m, 2H, –CH2), 1.25 (m, 4H, –CH2); 13C NMR (125 MHz, D2O): δ = 141.48, 135.79, 131.13, 128.94, 125.40, 122.74, 120.98, 101.49, 83.19, 73.78, 71.96, 71.31, 70.15, 59.97, 47.46, 36.29, 34.83, 31.04, 28.73, 23.17, 20.47; ESI-MS (m/z): calculated for [M+] 1284.51, found 1284.66; Anal. Calcd for C58H93O37N3S: C 47.83, H 6.44, N 2.89; found: C 47.85, H 6.42, N 2.86.
Mono-6A-[3-(2-ammonioethyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin chloride (7a)
A solution of 6a (2.80 g, 2 mmol) in water (10 mL) was added on the top of an Amberlite IRA-900 (Cl) resin column to convert tosylate into chlorine. The anion exchange lasted for 12 h before collecting the eluent. The eluent was concentrated to 2 mL before adding dropwise into dilute hydrochloric acid solution (0.1 mM, 5 mL). The crude product was precipitated out of acetone (40 mL). A recrystallization from water afforded the final product 7a (1.66 g, 64%) as a white solid: d.p. ∼224.2 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.02 (s, 1H,CH-2im), 7.75 (s, 1H, CH-4im), 7.70 (s, 1H, CH-5im), 5.99 (br, 1H, OH-2CD), 5.88–5.61 (m, 13H, OH-2CD and OH-3CD), 4.99–4.73 (br, 7H, H-1CD), 4.62–4.38 (br, 6H, OH-6CD), 4.32 (br, 1H, H-6′CD), 4.20–4.10 (br, 1H, H-3′CD), 4.02–3.92 (br, 1H, H-5′CD), 3.82 (br, 2H, –CH2-N-im), 3.71–3.48 (m, 24H, H-5CD, H-3CD and H-6CD), 3.482–3.15 (m, 12H, H-2CD and H-4CD), 3.11–3.05 (m, 1H, H-4′CD), 3.02 (br, 2H, –CH2-N-ammnium), 2.90–2.84 (m, 1H, H-2′CD); 13C NMR (125 MHz, D2O): δ = 134.43, 123.22, 121.17, 102.13, 83.07, 73.53, 72.51, 71.25, 60.27, 34.04, 26.47; ESI-MS (m/z): calculated for [M+] 1229.12, found 1228.56; Anal. Calcd for C48H81Cl2O34N3: C 43.84, H 6.21, N 3.20; found: C 43.89, H 6.22, N 3.16.
Mono-6A-[3-(3-ammoniopropyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin chloride (7b)
White solid (yield 68%): d.p. ∼217.0 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.17 (s, 1H,CH-2im), 7.81 (s, 1H, 1H, CH-4im), 7.74 (s, 1H, CH-5im), 5.68–5.84 (m, 13H, OH-2CD and OH-3CD), 4.97 (br, 1H, H-1CD), 4.82–4.85 (br, 6H, H-1CD), 4.27–4.48 (br, 6H, OH-6CD), 4.14 (br, 1H, H-3′CD), 3.99 (br, 1H, H-5′CD), 3.86 (br, 2H, –CH2-N-im), 3.55–3.65 (m, 25H, H-5CD, H-3CD and H-6CD), 3.28–3.47 (m, 12H, H-2CD and H-4CD), 3.21 (m, 1H, H-4′CD), 3.06 (m, 1H, H-2′CD), 2.82 (br, 2H, –CH2-N-ammnium), 2.06 (m, 2H, –CH2); 13C NMR (125 MHz, D2O): δ = 132.38, 126.27, 122.64, 102.53, 83.03, 73.85, 72.55, 72.06, 71.11, 60.72, 37.44, 29.27, 24.67; ESI-MS (m/z): calculated for [M+] 1242.46, found 1242.59; Anal. Calcd for C49H83Cl2O34N3: C 44.28, H 6.29, N 3.16; found: C 44.33, H 6.31, N 3.13.
Mono-6A-[3-(4-ammoniobutyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin chloride (7c)
White solid (yield 54%): d.p. ∼195.5 °C; 1H NMR (500 MHz, D2O): δ = 7.80 (s, 1H,CH-2im), 7.54 (s, 1H, 1H, CH-4im), 7.49 (s, 1H, CH-5im), 4.37–3.87 (br, 7H, H-1CD), 4.37–3.87 (br, 1H, H-3′CD), 3.77–3.73 (br, 1H, H-5′CD), 3.67 (br, 2H, –CH2-N-im), 3.61–3.31 (m, 26H, H-5CD, H-3CD and H-6CD), 3.31–3.06 (m, 12H, H-2CD and H-4CD), 3.06–3.00 (m, 1H, H-4′CD), 2.98–2.88 (br, 1H, m, 1H, H-2′CD), 2.69 (t, 2H, J = 6.0 Hz, –CH2-N-ammnium), 1.65 (q, 2H, J = 6.0 Hz, –CH2), 1.37 (q, 2H, J = 6.0 Hz, –CH2); 13C NMR (125 MHz, D2O): δ = 136.37, 126.54, 123.62, 101.92, 82.24, 73.51, 72.19, 71.53, 60.52, 38.84, 34. 56, 29.27, 22.87; ESI-MS (m/z): calcd for [M+] 1256.48, found 1256.38; Anal. Calcd for C50H85Cl2O34N3: C 44.71, H 6.38, N 3.13; found: C 44.77, H 6.42, N 3.12.
Mono-6A-[3-(6-ammoniohexyl)-imidazol-1-ium]-6A-deoxy-β-cyclodextrin chloride (7d)
White solid (yield 57%): d.p. ∼190.8 °C; 1H NMR (500 MHz, DMSO-d6): δ = 9.18 (s, 1H,CH-2im), 7.83 (s, 1H, 1H, CH-4im), 7.76 (s, 1H, CH-5im), 5.95 (br, 1H, OH-2CD), 5.85–5.56 (m, 13H, OH-2CD and OH-3CD), 4.97 (br, 1H, H-1′CD), 4.83–4.70 (br, 6H, H-1CD), 4.53–4.38 (br, 6H, OH-6CD), 4.42–4.32 (br, 2H, H-6′CD), 4.18 (br, 2H, –CH2-N-im), 3.97 (br, 1H, H-3′CD), 3.86 (br, 1H, H-5′CD), 3.73–3.56 (m, 24H, H-5CD, H-3CD and H-6CD), 3.48–3.22 (m, 12H, H-2CD and H-4CD), 3.06 (m, 1H, H-4′CD), 2.77 (br, 2H, –CH2-N-ammnium), 1.83 (br, 2H, –CH2), 1.56 (br, 2H, –CH2), 1.37 (br, 4H, 2-CH2), 1.27 (br, 2H, –CH2); 13C NMR (125 MHz, D2O): δ = 136.48 (C-2im), 123.26 (C-4im), 122.61(C-5im), 101.85 (C1), 81.59 (C4), 73.85 (C2), 72.55 (C3), 72.06 (C5), 71.11 (C5), 60.72(C6), 50.03 (C-N-im), 49.67 (C-N-ammnium), 37.44 (CH2), 29.05 (CH2), 26.56 (CH2), 25.06 (CH2); ESI-MS (m/z): calculated for [M+] 1285.22, found 1284.69; Anal. Calcd for C52H89Cl2O34N3: C 45.55, H 6.54, N 3.06; found: C 45.58, H 6.56, N 3.01.
Mono-6A-[3-(3-imidazolpropyl)-ammonium]-6A-deoxy-β-cyclodextrin tosylate (9b)
A similar approach as for 5 was employed to afford the title compound as a white solid (82%): d.p. ∼210 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.02 (s, 1H,CH-2im), 7.95 (s, 1H, CH-4im), 7.63 (s, 1H, CH-5im), 7.18 (d, 2H, J = 9.0 Hz, CH-ortho-OTs), 6.88 (d, 2H, J = 9.0 Hz, CH-meta-OTs), 5.70–5.85 (m, 14H, OH-2CD and OH-3CD), 4.98 (br, 1H, H-1CD), 4.83–4.97 (br, 6H, H-1CD), 4.43–4.57 (br, 6H, OH-6CD), 3.96 (t, 2H, –CH2-N-im), 3.55–3.63 (m, 21H, H-5CD, H-3CD and H-6CD), 3.28–3.41 (m, 12H, H-2CD and H-4CD), 3.21 (m, 1H, H-4′CD), 3.06 (m, 1H, H-2′CD), 3.02 (br, 2H, –CH2-N-ammnium), 2.22 (s, 3H, –CH3-OTs), 1.83 (m, 2H, –CH2); ESI-MS (m/z): calculated for [M+] 1242.46, found 1242.59; Anal. Calcd for C55H87O37N3S: C 46.71, H 6.20, N 2.97; found: C 46.72; H 6.25, N 2.93.
Mono-6A-[3-(3-imidazolpropyl)-ammonium]-6A-deoxy-β-cyclodextrin chloride (10b)
A similar approach as for 7 was used to afford the title product as a white solid (72%): d.p. ∼208 °C; 1H NMR (500 MHz, DMSO-d6) δ 7.61 (s, 1H,CH-2im), 7.14 (s, 1H, 1H, CH-4im), 6.85 (s, 1H, CH-5im), 5.69–5.84 (m, 14H, OH-2CD and OH-3CD), 4.97 (br, 1H, H-1CD), 4.83–4.87 (br, 6H, H-1CD), 4.43–4.57 (br, 6H, OH-6CD), 4.00 (t, 2H, –CH2-N-im), 3.99 (br, 1H, H-5′CD), 3.55–3.65 (m, 20H, H-5CD, H-3CD and H-6CD), 3.28–3.41 (m, 12H, H-2CD and H-4CD), 3.21 (m, 1H, H-4′CD), 3.06 (m, 1H, H-2′CD), 2.73 (br, 2H, –CH2-N-ammnium), 1.81 (m, 2H, –CH2); ESI-MS (m/z): calculated for [M+] 1242.46, found 1242.59; Anal. Calcd for C49H82ClO34N3: C 45.53, H 6.39, N 3.25; found: C 45.57; H 6.35, N 3.23.
Acknowledgements
We gratefully acknowledge the financial support from the Natural Science Foundation of Jiangsu Province (Grant No. BK2010486) and the Research Fund for the Doctoral Program of Higher Education of China (Grant No. 20113219110037). The authors would like to thank Lan Yi (Center of Analysis and Characterization, Nanjing University of Science and Technology) for NMR measurements.References
- J. Szejtli, T. Osa (eds), Comprehensive Supermolecular Chemistry, Vol. 3, Cyclodextrins, Pergamon, Oxford 1997 Search PubMed.
- W. H. Tang and S. C. Ng, Nat. Protoc., 2008, 3, 691 CrossRef CAS.
- Z. Juvancz, R. Kendrovics, R. Lvanyi and L. Szente, Electrophoresis, 2008, 29, 1701 CrossRef CAS.
- (a) H. Lu and G. Chen, Anal. Methods, 2011, 3, 488 RSC; (b) S. Fanali, Electrophoresis, 2009, 30, S203 CrossRef.
- (a) R.-Q. Wang, T. T. Ong and S. C. Ng, J. Chromatogr., A, 2008, 1203, 185 CrossRef CAS; (b) Y. Xiao, Y. Wang, T. T. Ong, L. Y. Ge, S. N. Tan, D. J. Young, T. T. Y. Tan and S. C. Ng, J. Sep. Sci., 2010, 33, 1797 CrossRef CAS; (c) T. T. Ong, R.-Q. Wang and S. C. Ng, J. Chromatogr., A, 2008, 1182, 136 CrossRef CAS.
- (a) T. de Boer, R. A. de Zeeuw, G. J. de Jong and K. Ensing, Electrophoresis, 2000, 21, 3220 CrossRef CAS; (b) V. Cucinotta, A. Contino, A. Giuffrida, G. Maccarrone and M. Messina, J. Chromatogr., A, 2010, 1217, 953 CrossRef CAS.
- (a) T. Schmitt and H. Engelhardt, Chromatographia, 1993, 37, 458 CrossRef; (b) O. H. J. Szolar, R. S. Brown and J. H. T. Luong, Anal. Chem., 1995, 67, 3004 CrossRef CAS; (c) Y. Tanaka, M. Yanagawa and S. Terabe, J. High Resolut. Chromatogr., 1996, 19, 421 CrossRef CAS.
- F. Wang and M. G. Khaledi, J. Chromatogr., A, 1998, 817, 121 CrossRef CAS.
- (a) W. H. Tang and S. C. Ng, Nat. Protoc., 2007, 2, 3195 CrossRef CAS; (b) W. H. Tang, I. W. Muderawan, S. C. Ng and H. S. O. Chan, Anal. Chim. Acta, 2006, 555, 63 CrossRef CAS; (c) W. H. Tang, I. W. Muderawan, T. T. Ong and S. C. Ng, Electrophoresis, 2005, 26, 3125 CrossRef CAS; (d) W. H. Tang, I. W. Muderawan, T. T. Ong and S. C. Ng, J. Chromatogr., A, 2005, 1091, 152 CrossRef CAS.
- (a) W. H. Tang, I. W. Muderawan, S. C. Ng and H. S. O. Chan, J. Chromatogr., A, 2005, 1094, 187 CrossRef CAS; (b) W. H. Tang, T. T. Ong, I. W. Muderawan and S. C. Ng, Anal. Chim. Acta, 2007, 585, 227 CrossRef CAS; (c) W. H. Tang and S. C. Ng, J. Sep. Sci., 2008, 31, 3246 CrossRef CAS.
- (a) H. Xiao and N. Cezar, J. Colloid Interface Sci., 2005, 283, 406 CrossRef CAS; (b) X. Jiang, Y. Qi, S. Wang and X. Tian, J. Hazard. Mater., 2010, 173, 298 CrossRef CAS.
- (a) C. O. Mellet, J. M. G. Fernández and J. M. Benito, Chem. Soc. Rev., 2011, 40, 1586 RSC; (b) C. Thiele, D. Auerbach, G. Jung, L. Qiong, M. Schneider and G. Wenz, Polym. Chem., 2011, 2, 209 RSC.
- (a) R. A. Glennon, N. A. Naiman, R. A. Lyon and M. Titelert, J. Med. Chem., 1988, 31, 1968 CrossRef CAS; (b) M. Buchholz, U. Heiser, S. Schilling, A. J. Niestroj, K. Zunkel and H. U. Demuth, J. Med. Chem., 2006, 49, 664 CrossRef CAS; (c) C. P. Bergstrom, C. P. Sloan and W. Y. Lau, Bioorg. Med. Chem. Lett., 2008, 18, 464 CrossRef CAS; (d) W. B. Wright, J. B. Press, P. S. Chan, J. W. Marsico, M. F. Haug, J. Lucas, J. Tauber and A. S. Tomcufick, J. Med. Chem., 1986, 29, 523 CrossRef CAS.
- (a) G. Galaverna, R. Corradini, A. Dossena and R. Marchelli, Electrophoresis, 1999, 20, 2619 CrossRef CAS; (b) G. Galaverna, R. Corradini, A. Dossena, R. Marchelli and G. Vecchio, Electrophoresis, 1997, 18, 905 CrossRef CAS.
- F. Lelièvre, P. Gareil and A. Jardy, Anal. Chem., 1997, 69, 385 CrossRef.
- F. Wang and M. G. Khaledi, J. Chromatogr., A, 1998, 817, 121 CrossRef CAS.
- T. T. Ong, W. H. Tang, I. W. Muderawan, S. C. Ng and H. S. O. Chan, Electrophoresis, 2005, 26, 3839 CrossRef CAS.
- L. Prokai, R. Ramanathan, J. Nawrocki and J. Eyler, J. Incl. Phenom. Mol. Recognit. Chem., 1996, 25, 117 CrossRef CAS.
- (a) W. H. Tang, S. C. Ng and H. S. O. Chan, J. Inclusion Phenom. Macrocyclic Chem., 2006, 56, 287 CrossRef CAS; (b) P. Suresh and K. Pitchumani, J. Org. Chem., 2008, 73, 9121 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Instrumentation; detailed synthesis of 3, 4, 8a and 9a; NMR and ESI-MS spectra for 5–7. See DOI: 10.1039/c2ra20086d/ |
| ‡ Jie Zhou and Yun Dai contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2012 |