Immobilization of ionic liquids in θ-zirconium phosphate for catalyzing the coupling of CO2 and epoxides
Hang
Hu
ab,
Jarett C.
Martin
a,
Meng
Zhang
b,
Cara S.
Southworth
a,
Min
Xiao
b,
Yuezhong
Meng
*b and
Luyi
Sun
*a
aDepartment of Chemistry and Biochemistry & Materials Science and Engineering Program, Texas State University-San Marcos, San Marcos, TX 78666, USA. E-mail: luyi.sun@txstate.edu; Fax: (512) 245-2374; Tel: (512) 245-5563
bThe Key Laboratory of Low-carbon Chemistry & Energy Conservation of Guangdong Province, State Key Laboratory of Optoelectronic Materials and Technologies, School of Physics and Engineering, Sun Yat-sen University, Guangzhou, 510275, P. R. China. E-mail: mengyzh@mail.sysu.edu.cn; Fax: (0086) 20-8411-4113; Tel: (0086) 20-8411-4113
First published on 13th February 2012
Abstract
The immobilization of 1-butyl-3-methylimidazolium chloride (BMIMCl) was achieved via the intercalation of BMIMCl into layered θ-zirconium phosphate (θ-ZrP), a hydrated form of α-zirconium phosphate. The intercalation process was studied by controlling the feed ratio of BMIMCl to θ-ZrP. X-ray diffraction characterization revealed that the formed intercalation compounds exhibited multiple phases depending on the intercalation ratio. θ-ZrP served as an excellent host for the intercalation of BMIMCl. The immobilized BMIMCl was evaluated via a coupling reaction of CO2 and propylene oxide to synthesize propylene carbonate. The results showed that the immobilized BMIMCl could maintain a similar catalytic reactivity to un-supported BMIMCl. After immobilization, the BMIMCl catalyst could be effectively separated from the products and recycled, and the recycled catalyst maintained high reactivity. Overall, the immobilization of BMIMCl into θ-ZrP enables BMIMCl to be a “greener” catalyst for a green chemical process.
Introduction
Ionic liquids (ILs) have attracted significant interest due to their unique properties and wide applications.1–4 The earlier stage of research focused on ILs as designer “green” solvents (including their low volatility, immiscibility with regular organic/inorganic solvents and facile property tunability) for various applications.1,5 In the last decade, the application of ILs has been significantly broadened to many new fields,6 such as catalysis,7–9 electrolytes,10 lubricants,11 biomass processing,12–16 energetic materials,17etc. While some “green” features of ILs (such as being nontoxic, nonflammable and nonvolatile) have proved to be not the case for certain ILs,5 most ILs continuously play a key role in green chemistry. Many new green applications of ILs, such as serving as a solvent for lignocellulose in biomass processes12–16 and as a catalyst for CO2 fixation reactions, have shown to be very promising.While certain ILs have proven to be effective in catalyzing coupling reactions to convert CO2 to valuable chemicals,18–20 two major drawbacks, cost and viscosity, prevent their practical applications. One of the most promising approaches to solve these two problems is to immobilize ILs into solid supports.21–23 In fact, immobilization of ILs can not only help reduce cost and avoid the viscosity issue, but also enhance catalytic efficiency, facilitate catalyst separation and recycling, and even bring out new applications.24,25 For example, the immobilization of ILs into solid supports may result in a delayed release effect, or controllable release.26 In brief, after immobilization, ILs may perform better and turn out to be “greener” in terms of catalyst separation and recycling.
Porous silica and zeolites have been used as IL supports.9,27,28 Another group of supports that have been explored are layered compounds.29–31 The packing of parallel layers creates interlayer space. Ions or molecules that are larger than the interlayer gap may not be able to diffuse between the layers freely. However, 2-dimensional layers are not as rigid as their three-dimensional counterparts. If extra energy is supplied, the layers may be spread apart and the guest molecules can be immobilized within the galleries.32,33 In fact, in this way the immobilized guests can be better protected and the release of guests from the interlayer space might also be controlled, which would be beneficial for certain applications. The intercalation processes are usually driven by a reaction (acid–base, oxidation–reduction, or coordination reaction),34–36 ultrasonication,37 or microwave radiation.38 One of the most widely investigated layered compounds is α-zirconium phosphate (α-ZrP), Zr(HPO4)2·H2O. α-ZrP is an acidic inorganic ion exchanger, which exhibits characteristics such as high ion exchange capacity, highly ordered structure, ease of synthesis and ease of crystallinity and size control.39–46 It has been used as a model system for intercalation chemistry studies,43,44,46–48 as well as in a wide range of functional materials.49–59
Several successful approaches have been developed for the intercalation of bulky molecules into a α-ZrP framework.34,60–64 The guest molecules range from metal ions to macromolecules.65–67 However, previous attempts to directly intercalate bulky imidazolium-based ILs into α-ZrP in aqueous solution were not successful.68 Alternatively, intercalation of imidazolium-based ILs into α-ZrP was managed after pre-intercalating α-ZrP with butylamine.68 However, the pre-intercalated amines may significantly poison a wide range of catalysts. Hu et al. recently reported a mechanochemical approach to immobilize ILs into α-ZrP without using any solvent.69 However, it is highly desirable to explore the immobilization of ILs in layered compounds via a regular solution approach without pre-intercalation, which can be readily adopted by industrial chemical processes.
Colón and coworkers have developed a systematic methodology to take advantage of a hydrated form of α-ZrP, θ-ZrP, as the host to intercalate bulky molecules, such as insulin and luminescent metal complexes.63,64,67,70–77 θ-ZrP was first observed by Clearfield et al. in 1969.78 Kijima et al. reported a method to directly synthesize θ-ZrP in 1982.79 θ-ZrP contains approximately six hydration water molecules and exhibits a wider interlayer distance of ca. 10.3–10.4 Å,70,79,80 in contrast to 7.6 Å for α-ZrP. The slight variation in the interlayer distance of θ-ZrP originates from the minor deviation in the number of hydration water molecules in θ-ZrP. The increased interlayer distance of θ-ZrP significantly facilitates intercalation.70,79 After dehydration, θ-ZrP converts to the α-phase and the intercalation compounds maintain all of the physical and chemical properties as if α-ZrP were used as the original host. Therefore, θ-ZrP acts as a more ideal host for intercalation reactions, particularly for those large species that are difficult to directly intercalate into α-ZrP due to their size mismatch. Herein, we report the intercalation of ILs into the θ-ZrP layered structures in an aqueous dispersion and the evaluation of the immobilized IL as a catalyst for CO2 fixation. The main goal is to immobilize the IL to be a “greener” catalyst for green chemical applications.
Experimental
Zirconyl chloride octahydrate (ZrOCl2·8H2O, 98%), phosphoric acid (85%) and 1-butyl-3-methylimidazolium chloride (BMIMCl) were purchased from Sigma-Aldrich. Ethanol was obtained from EMD. All chemicals were analytical reagent grade and were used as received. Propylene oxide (PO) with a purity of 95.0% was pretreated with potassium hydroxide and refluxed over calcium hydride for 24 h. It was subsequently distilled under dry nitrogen gas and stored over 4A molecular sieves prior to use. CO2 (99.99%) was purchased from Shanghai Industry Gases Co. Ltd.θ-ZrP was synthesized following procedures reported by Marti et al.70 In a 500 mL flask, 100 mL of 0.05 M ZrOCl2 aqueous solution was refluxed with 100 mL of 6.0 M H3PO4 aqueous solution at 94 °C for 48 h. The product was rinsed with deionized water and ethanol and then collected by centrifugation. Under sonication, the synthesized θ-ZrP can be well-dispersed in water to form a uniform suspension. To a pre-determined amount of θ-ZrP suspension, BMIMCl was added, followed by sonication treatment for 1 h using a sonication bath (Branson, Model 5510). Various amounts of BMIMCl were reacted with θ-ZrP and the “intercalation ratio” was defined by the available cations in BMIMCl to the total cation exchange capacity of θ-ZrP in the initial formulation. For example, θ-ZrP/BMIMCl-200 refers to a sample prepared via the above procedures using starting materials formulated with an amount of BMIMCl that counts as 200% of the total exchangeable cations in θ-ZrP.
X-ray diffraction (XRD) patterns were recorded using a Bruker D8 diffractometer with Bragg–Brentano θ–2θ geometry (20.0 kV and 5.0 mA), using a graphite monochromator with Cu-Kα radiation. The XRD patterns of the intercalation compounds were measured in their dry form. Prior to each XRD characterization, a prepared intercalation compound aqueous dispersion was cast on a clean silicon wafer and then dried under ambient conditions overnight. For comparison, both dehydrated θ-ZrP (dried the same as intercalation compounds) and hydrated θ-ZrP were characterized as controls.
The thermal stability of the intercalation compounds was characterized by a thermogravimetric analyzer (TGA, TA Instruments Model Q50) under an air flow (60 mL min−1) at a heating rate of 10 °C min−1. Prior to each test, all samples were briefly dried under ambient conditions for 24 h and were then pre-heated to 90 °C for 30 min to remove the absorbed moisture in an air flow directly in TGA, subsequently cooled down to 50 °C and finally the heating history from 50 to 800 °C was recorded.
The catalysis evaluation of the immobilized BMIMCl was carried out through a coupling reaction of CO2 and PO20 in a 100 mL stainless steel autoclave equipped with a mechanical stirrer. For a typical reaction process, the immobilized BMIMCl and PO were charged into the reactor, which was pressurized with CO2 to 3.0 MPa, and reacted at 110 °C for 12 h. The reactor was then cooled to room temperature. The immobilized catalyst precipitated and was subsequently separated from the product and un-reacted PO, which formed a homogenous liquid phase. The un-reacted PO was separated by the distillation of the liquid mixture under vacuum, and the product propylene carbonate was collected. The recovered catalyst was further evaluated under the same reaction conditions.
Results and discussion
θ-ZrP was synthesized following the procedures described by Marti et al.70 The interlayer distance of the synthesized θ-ZrP was characterized by XRD and an interlayer distance of 10.4 Å was observed, which is in agreement with the previous reports.80,81The direct intercalation of BMIMCl was carried out in an aqueous dispersion. Fig. 1 presents the XRD patterns of θ-ZrP/BMIMCl intercalation compounds at various BMIMCl to θ-ZrP ratios. After drying, most of the hydration water in θ-ZrP was removed and, thus, the interlayer distance was reduced from 10.4 to 7.6 Å, which is identical to the interlayer distance of α-ZrP and consistent with the literature.70,78 But the peak at 7.6 Å is absent in the XRD patterns of all of the θ-ZrP/BMIMCl intercalation compounds, except that of θ-ZrP/BMIMCl-5, which only shows a brief shoulder at ca. 7.6 Å. This result indicates that effective intercalation has occurred between θ-ZrP and BMIMCl, leaving no pristine phase of θ-ZrP, even at a very low BMIMCl loading level of 10% (θ-ZrP/BMIMCl-10). Meanwhile, new intensive peaks located at lower 2θ degrees were observed. The initial characterization results have already demonstrated the advantage of θ-ZrP as a host for intercalation over α-ZrP, which cannot be intercalated by BMIMCl directly.68
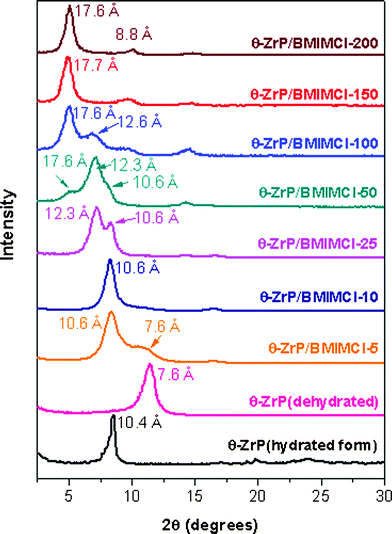 | ||
| Fig. 1 XRD patterns of pristine θ-ZrP and θ-ZrP/BMIMCl intercalation compounds with various BMIMCl loadings. | ||
As shown in Fig. 1, the interlayer distance of the intercalation compounds increased with an increasing loading of BMIMCl being intercalated with the assistance of ultrasonication. In some cases, multiphase intercalation compounds formed. When the intercalation ratio was as low as 5%, a phase with an interlayer distance of 10.6 Å formed. But a brief shoulder at 7.6 Å still remained, which indicated the existence of a tiny portion of un-intercalated θ-ZrP, which was converted to α-ZrP after dehydration. At 10% intercalation ratio, this shoulder at 7.6 Å completely disappeared, suggesting the elimination of pristine θ-ZrP phase. When the intercalation ratio reached 25%, besides the 10.6 Å peak, a new peak corresponding to an interlayer distance of ca. 12.3 Å formed. When the intercalation ratio was raised to 50%, the intensity ratio of the two peaks at 12.3 and 10.6 Å increased. The phase at ca. 12.3 Å turned out to be much more dominating. Meanwhile, a new shoulder at ca. 17.6 Å appeared. When the intercalation ratio was further increased to 100%, the intercalation compound with an interlayer distance of 17.6 Å became the main phase of θ-ZrP/BMIMCl-100. As the intercalation ratio kept rising (θ-ZrP/BMIMCl-150 and θ-ZrP/BMIMCl-200), only one diffraction peak at ca. 17.6 Å was observed, which indicates that all of the phases were finally converted to the 17.6 Å phase. During the entire process, three phases of intercalation compounds with interlayer distances of 10.6 Å, 12.3 Å and 17.6 Å, respectively, formed.
The phase transition behavior with the increasing intercalation ratio is believed to be due to the orientation of guest molecules.44,47,48 Similar intercalation phase transition processes have been reported in the literature and a model of possible orientations of guest molecules was proposed.63 When the intercalation ratio was low, the peak at 10.6 Å indicates a 4.0 Å interlayer distance expansion, since the zirconium phosphate layer thickness is 6.6 Å.63 Considering that the dimensions of BMIM+ are roughly 2.9 × 6.6 × 10.8 Å3,69 the 2.9 Å thickness is in reasonable agreement with the observed expansion in the interlayer distance, because at low intercalation ratios, hydration water molecules within θ-ZrP usually cannot be completely displaced.63 The remaining hydration water molecules within the layers (which can also be verified by the following TGA results, as shown in Fig. 2) typically lead to an additional slight increase in interlayer distance. In this phase BMIM+ cations were virtually parallel to the θ-ZrP layers. As the BMIMCl loading increased, in order to accommodate more BMIM+ in the θ-ZrP gallery, the alignment of BMIM+ changed from parallel to a tilted angle. Two possible arrangements of BMIM+ are proposed based on geometry calculations: a 56° tilted angle of the 6.6 Å molecular axes with respect to the layers plus a trace amount of hydration water molecules result in an interlayer distance of 12.3 Å and a double-layer of BMIM+ at angle tilted of 56° leads to an interlayer distance of 17.6 Å. The reason that BMIM+ cations prefer to tilt to 56° is probably due to the interlayer structure of θ-ZrP and the geometry of BMIM+ cations. It should be noted that the interlayer space of θ-ZrP is not flat. The P-OH Brønsted acid groups point inside the gallery and the ones on the top layer are not located exactly above the ones on the bottom layer.48 Such an interlayer geometric structure probably forces BMIM+ cations to tilt to 56° and fit themselves into the zigzag gallery spacing, which requires the lowest energy compared to other tilt angles.
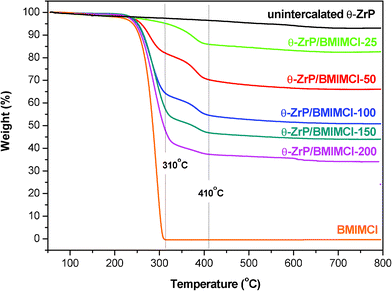 | ||
| Fig. 2 TGA thermograms of BMIMCl, un-intercalated θ-ZrP and θ-ZrP/BMIMCl intercalation compounds with various BMIMCl loadings. | ||
The TGA thermograms of neat θ-ZrP, BMIMCl and θ-ZrP/BMIMCl intercalation compounds with different BMIMCl loadings are shown in Fig. 2. Prior to each test, the samples were isothermed at 90 °C for 30 min in an air flow to remove absorbed moisture, then cooled down to 50 °C to start the TGA test. For θ-ZrP, the first weight loss that occurred between 140 and 170 °C corresponds to the removal of hydration water.48 Its weight loss after 450 °C can be accounted for by the decomposition of Zr(HPO4)2 to ZrP2O7.48 BMIMCl was decomposed and lost all the weight in the temperature range 230–310 °C. Decomposition of θ-ZrP/BMIMCl intercalation compounds took place mainly in four temperature regions at ca. 140–170, 220–310, 310–410 and 450–680 °C. The first weight loss is attributed to the removal of hydration water molecules. The second weight loss corresponds to the degradation of BMIMCl adsorbed on the θ-ZrP surface. With the increasing loading level of BMIMCl, the concentration of the adsorbed BMIMCl on θ-ZrP/BMIMCl intercalation compounds’ surfaces increased, which agrees well with the amount of the second weight loss of the intercalation compounds. The third weight loss can be assigned to the degradation of the intercalated BMIMCl in the θ-ZrP gallery. Compared with neat BMIMCl, which was completely decomposed before 310 °C, the delayed degradation of BMIMCl is believed to be due to the protection by θ-ZrP inorganic layers. It should be noted that the dividing line of 310 °C is defined according to the ending degradation temperature of neat BMIMCl. For the intercalation compounds with a high concentration of BMIMCl, such as θ-ZrP/BMIMCl-150 and 200, it appears that the ending decomposition temperature of the adsorbed BMIMCl is slightly higher than 310 °C according to their TGA thermograms. This phenomenon is believed to be due to the different distribution density of ILs near the edge in the θ-ZrP gallery. While it is widely accepted that the overall intercalation process starts from the edge of the layers and gradually progresses towards the center of the crystals,47,48,82 the ultrasonication applied appeared to help the guest molecules to diffuse deep into the layers after they are initially intercalated into the layer edge.44 This hypothesis is also supported by the elimination of neat θ-ZrP phase upon a very low concentration of BMIMCl (10%) being intercalated into θ-ZrP, as shown in Fig. 1. Thus, the intercalation compounds with a higher BMIMCl loading are expected to possess a higher density of BMIMCl in the edge region. Such BMIMCl near the layer edge was barely protected by θ-ZrP inorganic layers, exhibiting a similar thermal degradability to that of the adsorbed BMIMCl instead of the deeply intercalated ones, which are better protected and degrade at higher temperatures. The last weight loss of the θ-ZrP/BMIMCl intercalation compounds is due to the degradation of condensation water in θ-ZrP.
The immobilized BMIMCl in θ-ZrP was evaluated for catalysis applications using the following reaction.20
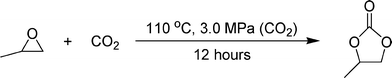
The emission of greenhouse gas CO2 has been increasing annually, which is a huge concern for global climate change. Hence, CO2 fixation has been of particular interest in the last few years.83,84 The synthesis of cyclic carbonates by CO2 and epoxide coupling reactions has been reported as a promising approach to convert CO2 to a valuable chemical.20 A variety of catalytic systems have been developed for the coupling reaction, including ionic liquids, which show high catalytic activity and selectivity.85–87 Nevertheless, the direct use of ILs will cause catalyst separation and recycling issues, as discussed above. The immobilization of ILs on a solid support is much more desirable, as it keeps the advantages of ILs and allows the immobilized ILs to serve as a heterogeneous catalyst. During the evaluation of the immobilized BMIMCl, no co-solvent or co-catalyst was used. The detailed reaction conditions and results are summarized in Table 1. While 0.7 g of neat BMIMCl led to a yield of 73.5%, 1.0 g of immobilized θ-ZrP/BMIMCl-200 (containing 0.7 g BMIMCl) resulted in a slightly higher yield of 78.9% under the same conditions. The minor improvement in yield is probably because the immobilized BMIMCl on θ-ZrP had a better dispersion in PO under stirring. After the first round of reaction, the immobilized catalyst was separated from the product and the unreacted PO and then used for a second reaction with fresh reactants. While it is difficult to separate the un-supported BMIMCl, which dissolves in the product, the immobilized θ-ZrP/BMIMCl-200 can be easily separated since it precipitated from the homogeneous liquid mixture of un-reacted PO and propylene carbonate. The recycled θ-ZrP/BMIMCl-200 exhibited close performance in terms of product yield to the previous reaction. The above results imply that the immobilized BMIMCl can maintain its catalytic efficiency as free BMIMCl. The immobilization can effectively facilitate the catalyst separation and recycling, while the recycled catalyst maintains reactivity. More detailed catalysis evaluation and optimization of such immobilized ILs for various new reactions are under way.
| Reaction | PO (mL) | CO2 (MPa) | T (°C) | Time (h) | Propylene carbonate yield (%) | ||
|---|---|---|---|---|---|---|---|
| 0.7 g BMIMCl (control) | 1.0 g θ-ZrP/BMIMCl-200 | ||||||
| 1 | Initial reaction | 15.0 | 3.0 | 110 | 12 | 73.5 | 78.9 |
| 2 | Recovered catalyst with fresh reactants | 15.0 | 3.0 | 110 | 12 | — | 63.4 |
While BMIMCl was used as an example for immobilization in θ-ZrP in this report, other ILs, including 1-hexyl-3-methylimidazolium chloride and 1-ethy-3-methylimidazolium acetate, have also been tried and successful immobilization was achieved. Thus, θ-ZrP has proven to be an ideal host for the immobilization of ILs. In addition to catalysis applications, θ-ZrP/IL intercalation compounds might also find applications in biosensors,88 batteries,89etc., in which ILs are preferred to be hybridized in a layered structure.
Conclusions
We have achieved direct immobilization of BMIMCl into θ-ZrP in solution state. Even at a very low intercalation ratio of 10%, the successful intercalation reaction was evidenced by both the XRD and TGA characterizations. As the intercalation ratio increased, a phase transition behavior was observed. θ-ZrP proved to be an excellent host for the intercalation of large molecules or ions. The θ-ZrP-immobilized BMIMCl was evaluated via a CO2 fixation reaction to synthesize propylene carbonate. The evaluation result showed that BMIMCl can maintain its catalytic reactivity after immobilization. Meanwhile, the immobilization could effectively facilitate the catalyst separation and recycling, and the recycled catalyst maintained high reactivity. The immobilization of BMIMCl into θ-ZrP renders BMIMCl a “greener” catalyst for a green chemical process.Acknowledgements
This research is sponsored by the Cottrell College Science Award from the Research Corporation for Science Advancement, the Key Laboratory of Low-Carbon Chemistry & Energy Conservation of Guangdong Province, and China High-Tech Development 863 Program (2009AA034900, 2009AA03Z340). L.S. would also like to acknowledge the U.S. Environmental Protection Agency, the U.S. Department of Agriculture (2011-38422-30803), and the Welch Foundation for partial support for this research. H.H. acknowledges the China Scholarship Council for offering her a scholarship to conduct research at Texas State University-San Marcos. We are grateful to Dr. Abraham Clearfield at Texas A&M University for valuable discussions.References
- T. Welton, Chem. Rev., 1999, 99, 2071–2083 CrossRef CAS.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182–1192 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- R. D. Rogers and G. A. Voth, Acc. Chem. Res., 2007, 40, 1077–1078 CrossRef CAS.
- T. L. Greaves and C. J. Drummond, Chem. Rev., 2007, 108, 206–237 CrossRef.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3691 CrossRef CAS.
- D. B. Zhao, M. Wu, Y. Kou and E. Min, Catal. Today, 2002, 74, 157–189 CrossRef CAS.
- C. P. Mehnert, Chem.–Eur. J., 2005, 11, 50–56 CrossRef.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621–629 CrossRef CAS.
- F. Zhou, Y. M. Liang and W. M. Liu, Chem. Soc. Rev., 2009, 38, 2590–2599 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS.
- M. B. Turner, S. K. Spear, J. D. Holbrey, D. T. Daly and R. D. Rogers, Biomacromolecules, 2005, 6, 2497–2502 CrossRef CAS.
- N. Sun, R. P. Swatloski, M. L. Maxim, M. Rahman, A. G. Harland, A. Haque, S. K. Spear, D. T. Daly and R. D. Rogers, J. Mater. Chem., 2008, 18, 283–290 RSC.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712–6728 CrossRef CAS.
- B. A. Simmons, S. Singh, B. M. Holmes and H. W. Blanch, Chem. Eng. Prog., 2010, 106, 50–55 CAS.
- M. Smiglak, W. M. Reichert, J. D. Holbrey, J. S. Wilkes, L. Y. Sun, J. S. Thrasher, K. Kirichenko, S. Singh, A. R. Katritzky and R. D. Rogers, Chem. Commun., 2006, 2554–2556 RSC.
- J. Sun, S.-i. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef CAS.
- L.-F. Xiao, F.-W. Li, J.-J. Peng and C.-G. Xia, J. Mol. Catal. A: Chem., 2006, 253, 265–269 CrossRef CAS.
- Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed., 1995, 34, 2698–2700 CrossRef CAS.
- C. P. Mehnert, E. J. Mozeleski and R. A. Cook, Chem. Commun., 2002,(24), 3010–3011 RSC.
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef CAS.
- C. M. Zhong, T. Sasaki, M. Tada and Y. Iwasawa, J. Catal., 2006, 242, 357–364 CrossRef CAS.
- G. J. Wang, N. Y. Yu, L. Peng, R. Tan, H. H. Zhao, D. H. Yin, H. Y. Qiu, Z. H. Fu and D. L. Yin, Catal. Lett., 2008, 123, 252–258 CrossRef CAS.
- S. C. Holmstrom, A. J. Patil, M. Butler and S. Mann, J. Mater. Chem., 2007, 17, 3894–3900 RSC.
- S. V. Sirotin, I. F. Moskovskaya, Y. G. Kolyagin, A. V. Yatsenko and B. V. Romanovsky, Russ. J. Phys. Chem. A, 2011, 85, 390–396 CrossRef CAS.
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC.
- Y. S. Ding, S. S. Wang, M. Zha and Z. G. Wang, Acta Phys.-Chim. Sin., 2006, 22, 548–551 CrossRef CAS.
- N. Jiang, H. Jin, Y. H. Mo, E. A. Prasetyanto and S. E. Park, Microporous Mesoporous Mater., 2011, 141, 16–19 CrossRef CAS.
- A. Riisager, R. Fehrmann, S. Flicker, R. van Hal, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2005, 44, 815–819 CrossRef CAS.
- J. M. Troup and A. Clearfield, Inorg. Chem., 1977, 16, 3311–3314 CrossRef CAS.
- M. S. Whittingham and A. J. Jacobson, Academic Press, 1982, p. 152.
- A. Clearfield and R. M. Tindwa, J. Inorg. Nucl. Chem., 1979, 41, 871–878 CrossRef CAS.
- J. W. Johnson, A. J. Jacobson, J. F. Brody and S. M. Rich, Inorg. Chem., 1982, 21, 3820–3825 CrossRef CAS.
- A. J. Jacobson, J. W. Johnson, J. F. Brody, J. C. Scanlon and J. T. Lewandowski, Inorg. Chem., 1985, 24, 1782–1787 CrossRef CAS.
- J. Z. Wang, Y. Hu, Y. Tang and Z. Y. Chen, Mater. Res. Bull., 2003, 38, 1301–1308 CrossRef CAS.
- U. Costantino, R. Vivani, V. Zima and L. Benes, Langmuir, 2002, 18, 1211–1217 CrossRef CAS.
- G. Alberti, Acc. Chem. Res., 1978, 11, 163–170 CrossRef CAS.
- A. Clearfield, J. Mol. Catal., 1984, 27, 251–262 CrossRef CAS.
- A. Clearfield, Annu. Rev. Mater. Sci., 1984, 14, 205–229 CrossRef CAS.
- C. V. Kumar, A. Bhambhani and N. Hnatiuk in Handbook of Layered Materials, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcel Dekker Inc., New York, 2004, pp. 313–372 Search PubMed.
- L. Sun, W. J. Boo, R. L. Browning, H.-J. Sue and A. Clearfield, Chem. Mater., 2005, 17, 5606–5609 CrossRef CAS.
- L. Sun, J. Y. O'Reilly, D. Y. Kong, J. Y. Su, W. J. Boo, H. J. Sue and A. Clearfield, J. Colloid Interface Sci., 2009, 333, 503–509 CrossRef CAS.
- L. Sun, W. J. Boo, H.-J. Sue and A. Clearfield, New J. Chem., 2007, 31, 39–43 RSC.
- W. J. Boo, L. Sun, J. Liu, A. Clearfield and H.-J. Sue, J. Phys. Chem. C, 2007, 111, 10377–10381 CAS.
- A. Clearfield in Progress in Intercalation Research, ed. W. Müller-Warmuth and R. Schollhorn, Kluwer, Dordrect, 1994, pp. 240–263 Search PubMed.
- A. Clearfield and U. Costantinoin Comprehensive Supramolecular Chemistry, ed. G. Alberti and T. Bein, Elsevier, Oxford, UK, 1996, pp. 107–149 Search PubMed.
- H.-J. Sue, K. T. Gam, N. Bestaoui, N. Spurr and A. Clearfield, Chem. Mater., 2004, 16, 242–249 CrossRef CAS.
- H.-J. Sue, K. T. Gam, N. Bestaoui, A. Clearfield, M. Miyamoto and N. Miyatake, Acta Mater., 2004, 52, 2239–2250 CrossRef CAS.
- L. Sun, W. J. Boo, D. Sun, A. Clearfield and H.-J. Sue, Chem. Mater., 2007, 19, 1749–1754 CrossRef CAS.
- W. J. Boo, L. Sun, J. Liu, A. Clearfield, H.-J. Sue, M. J. Mullins and H. Pham, Compos. Sci. Technol., 2007, 67, 262–269 CrossRef CAS.
- W. J. Boo, L. Sun, J. Liu, E. Moghbelli, A. Clearfield, H.-J. Sue, H. Pham and N. Verghese, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1459–1469 CrossRef CAS.
- W. J. Boo, L. Sun, G. L. Warren, E. Moghbelli, H. Pham, A. Clearfield and H.-J. Sue, Polymer, 2007, 48, 1075–1082 CrossRef CAS.
- L. Sun, W. J. Boo, A. Clearfield, H.-J. Sue and H. Q. Pham, J. Membr. Sci., 2008, 318, 129–136 CrossRef CAS.
- L. Sun, W.-J. Boo, J. Liu, A. Clearfield, H.-J. Sue, N. E. Verghese, H. Q. Pham and J. Bicerano, Macromol. Mater. Eng., 2009, 294, 103–113 CrossRef CAS.
- E. Moghbelli, L. Sun, H. Jiang, W. J. Boo and H.-J. Sue, Polym. Eng. Sci., 2009, 49, 483–490 CAS.
- L. Sun, J. Liu, S. R. Kirumakki, E. D. Schwerdtfeger, R. J. Howell, K. Al-Bahily, S. A. Miller, A. Clearfield and H.-J. Sue, Chem. Mater., 2009, 21, 1154–1161 CrossRef CAS.
- L. Sun and H.-J. Sue in Barrier Properties of Polymer Clay Nanocomposites, ed. V. Mittal, Nova Science Publishers, New York, USA, 2010, pp. 73–93 Search PubMed.
- U. Costantino, J. Chem. Soc., Dalton Trans., 1979,(2), 402–405 RSC.
- C. V. Kumar and Z. J. Williams, J. Phys. Chem., 1995, 99, 17632–17639 CrossRef CAS.
- C. V. Kumar and A. Chaudhari, J. Am. Chem. Soc., 2000, 122, 830–837 CrossRef CAS.
- E. J. Rivera, C. Figueroa, J. L. Colón, L. Grove and W. B. Connick, Inorg. Chem., 2007, 46, 8569–8576 CrossRef CAS.
- M. B. Santiago, G. A. Daniel, A. David, B. Casanas, G. Hernandez, A. R. Guadalupe and J. L. Colón, Electroanalysis, 2010, 22, 1097–1105 CAS.
- D. O’Hare, New J. Chem., 1994, 18, 989–998 Search PubMed.
- L. M. Liu, B. Shen, J. J. Shi, F. Liu, G. Y. Lu and J. J. Zhu, Biosens. Bioelectron., 2010, 25, 2627–2632 CrossRef CAS.
- A. Diaz, A. David, R. Perez, M. L. Gonzalez, A. Baez, S. E. Wark, P. Zhang, A. Clearfield and J. L. Colón, Biomacromolecules, 2010, 11, 2465–2470 CrossRef CAS.
- H. Y. Wang, M. J. Zou, N. Li and K. Li, J. Mater. Sci., 2007, 42, 7738–7744 CrossRef CAS.
- H. Hu, J. C. Martin, M. Xiao, C. S. Southworth, Y. Z. Meng and L. Y. Sun, J. Phys. Chem. C, 2011, 115, 5509–5514 CAS.
- A. A. Marti and J. L. Colón, Inorg. Chem., 2003, 42, 2830–2832 CrossRef CAS.
- R. A. Bermudez, Y. Colón, G. A. Tejada and J. L. Colón, Langmuir, 2005, 21, 890–895 CrossRef CAS.
- R. A. Bermudez, R. Arce and J. L. Colón, J. Photochem. Photobiol., A, 2005, 175, 201–206 CrossRef CAS.
- M. B. Santiago, M. M. Velez, S. Borrero, A. Diaz, C. A. Casillas, C. Hofmann, A. R. Guadalupe and J. L. Colón, Electroanalysis, 2006, 18, 559–572 CrossRef CAS.
- M. B. Santiago, C. Declet-Flores, A. N. D. Az, M. M. Velez, M. Z. Bosques, Y. Sanakis and J. L. Colón, Langmuir, 2007, 23, 7810–7817 CrossRef CAS.
- A. A. Marti, G. Paralitici, L. Maldonado and J. L. Colón, Inorg. Chim. Acta, 2007, 360, 1535–1542 CrossRef CAS.
- A. A. Marti, N. Rivera, K. Soto, L. Maldonado and J. L. Colón, Dalton Trans., 2007, 1713–1718 RSC.
- A. A. Marti and J. L. Colón, Inorg. Chem., 2010, 49, 7298–7303 CrossRef CAS.
- A. Clearfield, W. L. Duax, A. S. Medina, G. D. Smith and J. R. Thomas, J. Phys. Chem., 1969, 73, 3424–3430 CrossRef CAS.
- A. Kijima and Y. Fujio, Bulletin of the Japanese Society of Scientific Fisheries, 1982, 48, 1703–1709 CrossRef.
- G. Alberti, U. Costantino and J. S. Gill, J. Inorg. Nucl. Chem., 1976, 38, 1733–1738 CrossRef CAS.
- T. Kijima, Bull. Chem. Soc. Jpn., 1982, 55, 3031–3032 CrossRef CAS.
- A. Clearfield and S. P. Pack, J. Inorg. Nucl. Chem., 1975, 37, 1283–1290 CrossRef CAS.
- R. L. Paddock and S. T. Nguyen, J. Am. Chem. Soc., 2001, 123, 11498–11499 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009,(11), 1312–1330 RSC.
- D. J. Darensbourg, J. Lewis Samuel, J. L. Rodgers and J. C. Yarbrough, Inorg. Chem., 2003, 42, 581–589 CrossRef CAS.
- J. M. Sun, S. Fujita and M. Arai, J. Organomet. Chem., 2005, 690, 3490–3497 CrossRef CAS.
- J. A. Li, L. G. Wang, F. Shi, S. M. Liu, Y. D. He, L. J. Lu, X. Y. Ma and Y. Q. Deng, Catal. Lett., 2011, 141, 339–346 CrossRef CAS.
- Z. Dai, Y. Xiao, X. Yu, Z. Mai, X. Zhao and X. Zou, Biosens. Bioelectron., 2009, 24, 1629–1634 CrossRef CAS.
- T. E. Sutto, P. C. Trulove and H. C. De Long, Electrochem. Solid-State Lett., 2003, 6, A50–A52 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |