Single molecule diffusion on hard, soft and fluid surfaces†
Shengqin
Wang
,
Benxin
Jing
and
Yingxi
Zhu
*
Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: yzhu3@nd.edu
First published on 12th March 2012
Abstract
Molecular diffusion on a surface is often considered as a thermal energy-activated process of molecular hopping between adjacent adsorption sites on a surface and simply determined by molecule–surface interaction. In this work, we report distinct diffusive dynamics of probe molecules on methyl-terminated self-assembled monolayer (SAM), polymer brush layer and lipid bilayer by using fluorescence correlation spectroscopy (FCS) at a single-molecule level. We have observed that despite weaker molecule–surface interaction, the surface diffusion of probe molecules on soft polymer brush surface and fluid lipid bilayer can be much slower than that on hard SAM surface, suggesting a strong impact of interfacial dynamics of the underlying coating on molecular surface diffusion. To further examine the coupling of thermal activated molecular hopping and soft surface dynamics, we have investigated the diffusion of probe molecules on polymer brushes of varied grafting density and thickness, where the molecule–surface interaction remains nearly the same, yet the adopted conformations and dynamics of surface-grafted PNIPAM chains vary considerably; it is striking to observe that the diffusion of probe molecules could be further retarded on PNIPAM brush surfaces of lower grafting density or higher brush thickness, thereby exhibiting the presence of an optimal brush thickness range to facilitate fast surface diffusion of adsorbed probe molecules. All the observations combined lead to a general model by taking the dynamics of underlying surface layers into account to elucidate the molecular diffusion mechanism on varied surfaces.
Introduction
Surface diffusion of small molecules or colloids has attracted much scientific interest for decades because it plays a significant role in various technological fields from crystal and thin-film growth, surface chemical reaction and catalysis, to protein interaction and molecular motors.1–3 With the advancement of microfluidic devices and lab-on-chips, a fundamental understanding of how surface properties affect the diffusive dynamics of molecules at surfaces or interfaces becomes critically important to the design and performance of such micro/nano-fluidic devices, in which the surface effects could become much pronounced due to high surface-to-volume ratio and whose boundary effect can vary from the no-slip to the slip boundary condition with a strong dependence of surface treatment.4As previous studies mostly focus on the molecular diffusion on a hard solid surface, the mechanism of molecular diffusive dynamics on surface is generally considered as a process of molecular hopping between adjacent adsorption sites on a surface.5–7 The hopping sites are determined by attractive molecule–surface interaction, and the molecular motion on surface is thus activated by thermal fluctuation energy. Lately, a large variety of surface coatings have been employed to control molecule–surface interactions, including closely packed self-assembled monolayers (SAMs), adsorbed or grafted polymer thin films and even biological or biomimetic thin films. For instance, covalently surface-tethered poly(ethylene oxide) (PEO) or poly(N-isopropylacrylamide) (PNIPAM) thin films have been widely used to control protein and other biomolecule adsorption for various biomedical applications.8,9 Many of such functional surface coatings are not hard or in crystalline structures, but often soft or even mobile. Consequently, the diffusion of molecules on such surfaces can be drastically distinct from that on hard ones, because the hopping sites on surface are not statically fixed but dynamic. However, experimental study of the diffusive dynamics of adsorbed molecules on soft and mobile surfaces has been few,10–18 despite their broad technological applications as well as biophysical relevance to biomolecular transport through plasma membranes.19–21
In this work, we have systematically examined the surface diffusion of probe molecules on three types of contrasting surfaces, designated as hard, soft and fluid surfaces, based on their intrinsic interfacial dynamic characteristics. We select the commonly used methyl-terminated SAM as the representative hard surface, surface-tethered polymer brush thin film, which is a thin layer of brush-like polymer chains with one end attached to a substrate,21 as the representative soft surface, and lipid bilayer in its fluid phase as the representative fluid surface. Specifically, octadecyltriethoxylsilane (OTE) SAM, surface-tethered PNIPAM brush thin film and L-α-phosphatidylcholine (α-PC) lipid bilayer are strategically selected to be coated on a solid substrate and control similar interfacial interactions with probe molecules, thereby allowing us to study the effect of surface dynamic characteristics on molecular surface diffusion. We have employed fluorescence correlation spectroscopy (FCS) at a single molecule level to examine the surface diffusive dynamics of two small fluorescence probes, Rhodamine 6G (R6G) and Rhodamine 123 (R123), both of which can readily absorb on the three selected model surfaces by hydrophobic interaction. We have observed that despite the weakest attractive molecule–surface interaction, the surface diffusion of R6G on the fluid lipid bilayer appears the slowest, in sharp contrast to the fastest diffusion on OTE hard surface, despite the strongest hydrophobic attraction, suggesting a strong impact of interfacial dynamics of the underlying coating on molecule surface diffusion. A similar coupling effect is also observed with R123 on soft PNIAM brush layer and fluid α-PC lipid bilayer. Therefore, a general model based on the coupling of thermal activation and the dynamics of underlying surface layer is proposed to describe molecular diffusion on varied surfaces. To further examine the coupling effect, we have investigated the diffusion of probe molecules on PNIPAM brushes of varied grafting density and thickness, where the molecule–surface interaction remains nearly the same, yet the adopted conformations and dynamics of surface-grafted PNIPAM chains vary considerably; we have observed that measured surface diffusion coefficient of probe molecules shows a strong dependence of grafting density and thickness, exhibiting an optimal brush thickness range for fast surface diffusion of adsorbed probe molecules. This study of the coupling effect of thermal fluctuation and the dynamics of underlying surface layers on molecular surface diffusion provides important insights to molecular design of functional surface coatings for micro- and nano-fluidic devices, in which controllable diffusion and adsorption of molecules on surfaces are critical.
Experimental section
Materials
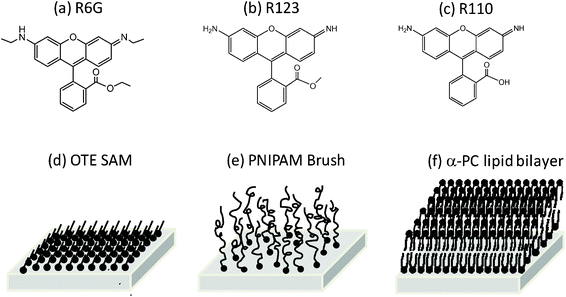 | ||
| Fig. 1 Chemical structures of (a) Rhodamine 6G, (b) Rhodamine 123 and (c) Rhodamine 110, and schematic drawings of three surfaces: (d) OTE monolayer, (e) PNIPAM brush and (f) α-PC lipid bilayer. | ||
To determine the PNIPAM brush thickness, PNIPAM brushes were also grafted from silicon wafers under exactly identical experimental conditions for ellipsometry characterization. The thickness of dry PNIPAM brushes (refractive index, n = 1.49) in air was determined by ellipsometry (Gaertner, model L116C) at a fixed laser wavelength of λ = 632.8 nm and a fixed incidence angle of 70°, while the wet thickness of swollen PNIPAM brushes immersed in deionized water was determined by phase-modulated ellipsometer (Beaglehole) with a custom-built fluid cell. The brush thickness was also confirmed by AFM (Veeco, Nanoscope IV) via the section analysis of scratches on PNIPAM brushes in air and in aqueous solutions using an AFM fluid cell. The grafting density of PNIPAM brushes was estimated from the ratio of the measured wet to dry brush thicknesses by using a mean-field theoretical mode as described in detail elsewhere.23 The LCST as well as surface hydrophobicity of PNIPAM brushes was determined by the static contact angle of water sessile drops using a goniometer (Rame–Hart, model 250) with a heating stage (INSTEC, model HCS60). The LCST of PNIPAM brushes of varied grafting density and thickness was found to be in a moderately narrow temperature range of 28–35 °C, across which the PNIPAM surface undergoes a transition from a hydrophilic surface to a hydrophobic one accompanied with PNIPAM chain collapse.
PNIPAM brushes of three different grafting densities, σ = 0.13, 0.33 and 0.62 chain nm−2, were synthesized and used in this work. At each grafting density, 5–7 different brush thicknesses were varied over the range of dry brush thickness, h = 0.8–30 nm by varying the polymerization time. For all synthesized PNIPAM brush surfaces using our LB-ATRP method, their molecular smoothness was confirmed by AFM, exhibiting that the measured root-mean-square (rms) roughness was less than 1.0 ± 0.2 nm over a scanning area of 10 μm ×10 μm in the ambient environment at T = 25 °C.23
Method
The diffusive dynamics of fluorescent probe molecules on varied surfaces was characterized by FCS at a single-molecule level.25–27 FCS was set up on an inverted microscope (Zeiss Axio A1) equipped with an oil-immersion objective lens (100×, NA = 1.4) as detailed elsewhere.27–29 Experimentally, probe molecules, R6G or R123 were first absorbed on the surface from its dilute aqueous solution of ∼5 nM over 10 min, and excess probe molecules were removed by gently exchanging the solution with deionized water (Barnstein Nanopure II). An argon laser (Melles Griot, λ = 488 nm) was well focused at the water-coating interface where the maximum in fluorescence photon counts was detected by two single photon counting modules (Hamamatsu); the laser focus at the aqueous interface, neither inside the fused silica substrate nor in the solution far above the substrate, was also confirmed with a presence of the smallest optical scattering spot exactly at the aqueous interface as observed by a CCD camera (Andor iXon). The emission fluorescence intensity, F(t) of probe molecules over a small focal volume, which was calibrated as ϖ ∼190 nm in the lateral diameter and z ∼3 μm in the vertical height by R6G in a dilute bulk solution, was measured to obtain the auto-correlation function, G(τ) = 〈δF(t)·δF(t + τ)〉/〈F(t)〉2 The surface diffusion coefficient, D as well as the surface concentration, [c] of probe molecules at varied surface coatings were thus obtained from G(τ) by fitting it with the two-dimensional Gaussian equation (eqn (1)):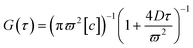 | (1) |
Results and discussion
We start with investigating the diffusive dynamics of a single probe molecule, R6G, at three distinct surfaces by FCS. Fig. 2(a) shows the normalized G(τ) by G(0) of R6G in a dilute bulk aqueous solution, on an OTE monolayer, a PNIPAM surface of dry brush thickness, h = 2.5 nm and grafting density, σ = 0.62 chain nm−2, and an α-PC lipid bilayer at T = 25 °C. For the case of R6G in a bulk solution, the obtained G(τ) is fitted with the three-dimensional Gaussian equation,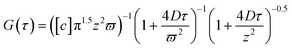 to obtain D = 280 μm2 s−1. For the cases of R6G diffusion on OTE, PNIPAM and lipid surface, the surface diffusion coefficient values, D = 10.9, 7.2, and 6.3 μm2 s−1, respectively, are obtained by fitting G(τ) with eqn (1). It is noted that the curve fittings for the cases of probe molecules on varied surfaces shown in Fig. 2 are very good, indicating that the molecular diffusion on surface is featured with normal diffusion as described by the single Fickian diffusion model to derive eqn (1).
to obtain D = 280 μm2 s−1. For the cases of R6G diffusion on OTE, PNIPAM and lipid surface, the surface diffusion coefficient values, D = 10.9, 7.2, and 6.3 μm2 s−1, respectively, are obtained by fitting G(τ) with eqn (1). It is noted that the curve fittings for the cases of probe molecules on varied surfaces shown in Fig. 2 are very good, indicating that the molecular diffusion on surface is featured with normal diffusion as described by the single Fickian diffusion model to derive eqn (1).
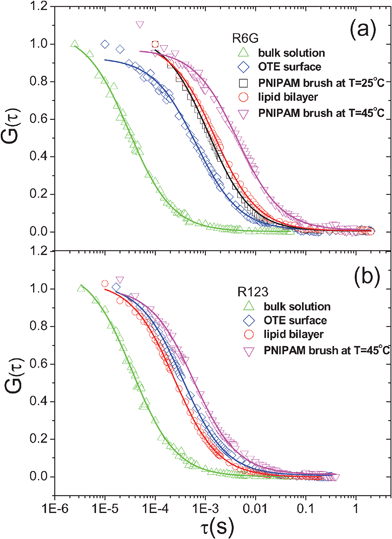 | ||
| Fig. 2 Normalized autocorrelation function, G(τ) by G(0) of (a) R6G and (b) R123 at T = 25 °C in bulk aqueous solution (△), on an OTE monolayer (⋄), on PNIPAM brush thin film of dry thickness, h = 2.5 nm and grafting density, σ = 0.62 chain nm−2 (□) as well as at T = 45 °C (▽), and on α-PC lipid bilayer (○). It is noted that at T = 25 °C, no adsorption of R123 on PNIPAM brush thin film of h = 2.5 nm and σ = 0.62 chain nm−2 is observed. | ||
It is evident that the dynamic process of R6G on surfaces considerably slows down from that in bulk solutions. Yet it is most interesting and surprising to observe that the slowest surface dynamics of R6G occurs on hydrophilic α-PC lipid bilayer, not on the most hydrophobic OTE surface with a water contact angle, θ = 110°, in stark contrast to the classical anticipation13 that the surface dynamics of adsorbed molecules is more retarded on a surface of stronger interfacial interaction; in other words, the apparent friction force experienced by R6G on soft, weakly attractive surfaces is higher than on hard, strongly attractive ones, as the apparent friction coefficient, ξ can be also estimated by using the Einstein equation with measured D as ξ = kT/D. With PNIPAM brushes, we have also compared the measured D at T = 25 °C, at which the surface is partially hydrophilic with θ = 55°, to that at T = 45 °C, at which the polymer surface becomes partially hydrophobic with θ = 72°, and observed the further slower surface dynamics of R6G with measured D = 1.8 μm2 s−1. Combined with all the observations, it appears to suggest that strong molecule–surface interaction can slow down the diffusion of molecules at surface, yet the molecular surface dynamics might be strongly coupled with the interfacial dynamics of underlying layers on a solid substrate, which could significantly retard the molecular motion at the interface.
To further verify the above observations, we have also examined the dynamics of R123 and R110 fluorescence probes on the same OTE, PNIPAM brush and α-PC lipid bilayer surfaces. For R110 that has the chemical structure highly similar to R123 but is more hydrophilic than R123 and R6G, no surface absorption is detected on all three surfaces, resulting in no measurable surface diffusion coefficient. For R123, as shown in Fig. 2b, the trend of surface diffusion coefficient on the three varied surfaces is somewhat different from that of R6G: No adsorption and consequently no diffusion is observed with R123 on PNIPAM brush at T < LCST with water contact angle, θ = 55°; however on α-PC lipid bilayer with θ of ∼2–5°, considerable adsorption of R123 is observed with a resulting measured D = 33.9 μm2 s−1. As is seen from the obtained D for R6G and R123 summarized in Table 1, it is clear that on each surface the diffusion of R123 is faster than that of R6G, suggesting the interaction of R123 probe with each surface is weaker than that of R6G; according to their chemical structures as shown in Fig. 1, it is reasonably deduced that the hydrophobic interaction contributes predominantly to the adsorption of R6G and R123 on these three surfaces. Additionally, it is worth noting that the contribution of electrostatic interaction to their surface adsorption, particularly on the amphiphilic α-PC lipid bilayer, is nearly excluded from the control experiments with a negatively charged dye, Alexa Fluor 488, with which no adsorption is observed on all three surfaces.
| OTE SAM (T = 25 °C) | PNIPAM brush (h = 2.5 nm, σ = 0.62 chain nm−2) | α-PC lipid bilayer (T = 25 °C) | ||
|---|---|---|---|---|
| T = 25 °C | T = 45 °C | |||
| Static water contact angle/° | 110 ± 1 | 55 ± 2 | 72 ± 2 | ∼2–530 |
| D R6G/μm2 s−1 | 10.9 ± 2.2 | 7.2 ± 1.1 | 1.8 ± 0.5 | 6.3 ± 0.7 |
| D R123/μm2 s−1 | 21.9 ± 2.7 | No adsorption | 13.6 ± 1.9 | 33.9 ± 2.4 |
Water contact angles on OTE, PNIPAM brush and α-PC lipid bilayer at T = 25 °C are measured to be θ = 110, 55 and ∼2–5°, respectively, to describe their surface hydrophobicity,30 indicating that the hydrophobic interaction strength and also the assumed resulting adsorption of R6G or R123 on the three surfaces is expected to fall in the order of OTE SAM > PNIPAM brush > α-PC lipid bilayer. On the contrary, the measured D of R6G shows the opposite trend with these three surfaces, that is DR6G-OTE > DR6G-PNIPAM > DR6G-lipid at T = 25 °C . It should be noted that at the same PNIPAM brush surfaces, the surface diffusion of R6G probe is further retarded at the PNIPAM surfaces of increased hydrophobicity by increasing solution T across its LCST as reported in our prior study.17
Therefore, all the results from measured D of R6G probes on different surfaces suggest that molecular surface diffusive dynamics is affected by both molecule–surface interaction and the dynamics of underlying surface layers. For solid surface coatings, such as OTE SAMs, the dynamics of underlying molecules are largely suppressed in the well-packed, ordered monolayer; as a result, the molecular surface diffusion can be exclusively determined by the interaction: strong molecule–surface interaction leads to slower diffusive dynamics due to higher activation energy as intuitively anticipated.31 In contrast, for fluid surface or soft polymer coatings, the dynamics of underlying surface can be comparable to that of surface-bound molecules; as a result, the measured surface diffusive dynamics can become strongly coupled with the dynamic fluctuation of these soft underlying coatings, exhibiting “tethered” surface diffusion.
Generally, in a simplest case when molecules walk on a solid, non-deformable surface, surface diffusion can be considered as a hopping process of the motion of molecules between adjacent adsorption sites. The hopping rate is determined by an attempt frequency, ν and the Boltzmann factor that is related to the thermal energy, kT and molecule–surface interaction, E to dictate the probability of an attempt leading to a successful surface hopping. The attempt frequency, ν is typically approximated to the vibrational frequency of the molecule, or the reciprocal of the characteristic time, τ0 of diffusive molecule in the bulk as 1/τ0.32 On a solid surface, the surface diffusion coefficient, D of a molecule can be simply estimated from its diffusion coefficient in the bulk, D0 and surface adsorption energy, E as eqn (2):
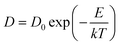 | (2) |
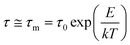 | (3) |
However, when the surface is not hard but soft and mobile, such as polymer brush or lipid bilayer surfaces due to their interfacial fluctuation of respective polymer chain segments or lipid molecule, the hopping sites on surfaces to accommodate molecular diffusion are not static but dynamic or even mobile. When the interaction between molecule and the mobile surface is strong and there is molecular adsorption on the surface, the diffusion of adsorbed molecules on such dynamic surfaces could be described as a coupled process between thermal activated dynamics of absorbed molecules with a characteristic time of τm, and the dynamics of an underlying mobile surface with a characteristic time of τs; if τs is comparable to τm, molecular surface diffusion can be greatly affected by the dynamics of surface. As the coupling is intrinsically dictated by the molecule–surface interaction, the overall characteristic time of molecular diffusion on dynamic surfaces can be approximately described as eqn (4):
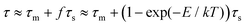 | (4) |
| τ = τm + (1 − (τ0/τm))τs | (5) |
In our recent work24 on the structural dynamics of lipid bilayers, we have investigated the dynamics of lipid bilayers in the fluid phase by mixing a fluorescent lipid probe, 1,2-dioleoyl-snglycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) ammonium (LRPE) with α-PC lipid at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :105. The lateral diffusion coefficient of α-PC lipid bilayer, Ds = 3.0 μm2 s−1 is obtained from the measured G(τ)/G(0) of LR-PE in uniform mixed lipid bilayer (see ESI†). Because the α-PC lipid molecule is rigid and in the fluid phase at T = 25 °C, the surface dynamics of α-PC lipid bilayer is entirely offered by its lateral mobility. Therefore, for α-PC lipid bilayer, τs ∼ 1/Ds. Eqn (5) thus can be reorganized as eqn (6):
:105. The lateral diffusion coefficient of α-PC lipid bilayer, Ds = 3.0 μm2 s−1 is obtained from the measured G(τ)/G(0) of LR-PE in uniform mixed lipid bilayer (see ESI†). Because the α-PC lipid molecule is rigid and in the fluid phase at T = 25 °C, the surface dynamics of α-PC lipid bilayer is entirely offered by its lateral mobility. Therefore, for α-PC lipid bilayer, τs ∼ 1/Ds. Eqn (5) thus can be reorganized as eqn (6):
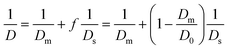 | (6) |
The critical dependence of f on interfacial interaction is also exhibited by comparing the trend of measured D of R6G on the three surfaces to that of R123. As summarized in Table 1, the measured D of R6G falls in the order of OTE SAM > PNIPAM brush > α-PC lipid bilayer, therefore the order of τ follows OTE SAM < PNIPAM brush < α-PC lipid bilayer, which is contrary to the order of τm resulted from interfacial interaction but intriguingly consistent with the order of τs contributed by underlying surface dynamics. By applying eqn (5), it is suggested that for R6G diffusion on lipid bilayer and PNIPAM brush, the surface dynamics plays a predominant role and the coupling degree f is significant due to strong surface interaction. In contrast, the order of measured τ of R123 on the three surfaces is not consistent with either the order of τm or that of τs, indicating that the contributions of both τm and τs to τ are comparable for R123 diffusion on lipid bilayer or PNIPAM brush. Because τs is intrinsically determined by the underlying surface, the weakened contribution of τs to τ in R123 surface diffusion clearly suggests a weaker coupling degree, which is resulted from the weaker surface interaction of R123 than R6G.
Alternatively, as indicated by eqn (5) or (6), we can also examine the dynamics of surface coatings of varied interfacial structure from the measured molecular surface diffusion, given that the molecule–surface interaction, E and the resulting Dm or τm essentially remains the same with same surface chemistry. Thus, to examine the effect of surface dynamics on molecular surface diffusion, we consider the surface-tethered polymer brushes as an ideal model system, whose interfacial dynamics can be effectively and easily modified by brush grafting density and thickness. With stimuli-responsive polymer brushes such as temperature-responsive PNIPAM brushes, their interfacial interaction with molecules can be also tuned considerably. Measured surface diffusion coefficients of R6G and R123 on two PNIPAM brush surfaces of h = 2.5 nm, σ = 0.62 chain nm−2 and h = 1.8 nm, σ = 0.33 chain nm−2 at varied T across the measured LCST are shown in Fig. 3. It is evident to see that the diffusive dynamics of R6G and R123 on PNIPAM brush surface slows down with enhanced probe molecule–PNIPAM hydrophobic interactions as T increases from 25 to 45 °C across its LCST ≈ 28–35 °C as shown in Fig. 3a,23 where PNIPAM brushes undergo a transition from hydrophilic and swollen brushes to a hydrophobic and collapsed thin film.
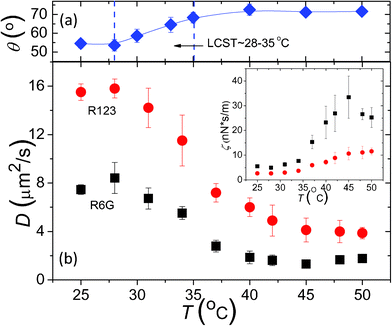 | ||
| Fig. 3 (a) Static water contact angle on PNIPAM brush surface of σ = 0.62 chain nm−2 and h = 2.5 nm, indicating the transition from a hydrophilic PNIPAM surface to a hydrophobic one as T increases across LCST ≈ 28–35 °C. (b) Measured surface diffusion coefficients, D of R6G (■) on PNIPAM surface of h = 2.5 nm and σ = 0.62 chain nm−2 and R123 (●) on PNIPAM surface of h = 1.8 nm and σ = 0.33 chain nm−2vs. solution temperature, T. Inset: Apparent friction coefficient, ζ that is inversely proportional to D is plotted vs. T. | ||
To investigate the molecular diffusion on PNIPAM brushes of varied h as well as the dependence of τs on h, we have systematically varied the brush thickness in air, h = 0.8–30 nm at varied σ = 0.13, 0.33 and 0.62 chain nm−2 and examined the diffusion of R6G on PNIPAM brushes at T = 25 °C.17 As shown in Fig. 4a, at constant σ = 0.62 chain nm−2 and T = 25 °C a non-monotonic h-dependence of R6G surface diffusion is intriguingly observed: the measured D is observed to first increase sharply (region I) to reach a plateau over the range of h = 1.7–7.1 nm (region II) and then decrease rapidly to a lower plateau at further increased h = 10–30 nm (region III). A similar trend is also observed at σ = 0.33 chain nm−2: at σ = 0.62 and 0.33 chain nm−2, an optimal brush thickness range is present with fast R6G surface diffusion, suggesting the weakest retarding effect of polymer brush dynamics on molecular surface diffusion. We have excluded the contribution of PINPAM surface roughness to the considerable difference in the measured diffusion coefficient of probes on PNIPAM brushes of varied thickness and density, whose molecular surface smoothness is verified experimentally23 with measured rms roughness, Rrms < 1.0 ± 0.2 nm over a scanning area of 10 μm × 10 μm. It is noted that at σ = 0.13 chain nm−2, the h-dependence is much less pronounced, which is possibly due to the slower diffusion of buried R6G molecules inside the PNIPAM brushes. The distance between two adjacent brush chains, s ≈ 2(πσ)−1/2 = 3.1 nm at σ = 0.13 chain nm−2 is much larger than the R6G molecular diameter, d = 0.8 nm, so that R6G molecules can penetrate and diffuse inside the layer of PNIPAM brushes at low σ due to low osmotic pressure of brush layer, which is also indicated by an increase of the concentration, [c] of absorbed R6G, measured by fitting G(τ) with eqn (1), with brush thickness nearly linear as shown in Fig. 4b. The penetration of tracer molecules into polymer brushes of relative low grafting density is also confirmed with the case of neutral polymer brush systems.18 However, at high σ, the possibility of burying R6G molecules inside the brushes is much reduced because it is energetically unfavored due to the high osmotic pressure of water-immersed polymer chains, which is also supported by the little variance in measured [c] of adsorbed R6G with h at σ = 0.33 and 0.62 chain nm−2 as shown in Fig. 4b. Additionally, in comparison between the measured D of R6G and R123 in a bulk solution and at the PNIPAM brush aqueous interfaces, the surface diffusive dynamics of both probe molecules on PNIPAM surfaces is found to be slowed down only by a factor of 5–10, much weaker than the expected dynamic retardation by 5–6 orders of magnitude for embedded molecules inside polymer network.15 We have also excluded the possible contribution of adsorbed probe molecules at the interface of PNIPAM brushes and their underlying initiator monolayer based on control experiments. The adsorption and diffusion of R6G on the underlying initiator monolayers of mixed BMTUP and MTUP was examined. It is found that the adsorption of R6G on initiator monolayers of varied MTUP coverage is much weaker than that on PNIPAM brushes, and the measured D of R6G on initiator monolayers is in a narrow range of 18–21 μm2 s−1, much greater than the reported D (= 0.3–7 μm2 s−1) here on PNIPAM brushes with a strong dependence of brush thickness. Hence, we strongly believe that the measured D is associated with adsorbed probe molecules on PNIPAM brush surfaces of high σ = 0.33 and 0.62 chain nm−2, not the ones buried inside the PNIPAM brush layer or at the PNIPAM-initiator monolayer interface.
![(a) Measured D of R6G on PNIPAM surfaces vs. h at varied σ = 0.62 (■), 0.33 (●) and 0.13 (▲) chain nm−2 at T = 25 °C. Dashed lines are used to indicate three distinct regions in observed surface dynamical behaviors at σ = 0.62 chain nm−2. Inset: Growth of PNIPAM brush thickness vs. ATRP time at σ = 0.62 (□) and 0.33 (○) chain nm−2. Dashed lines are used to indicate the critical ATRP time, above which the growth of bush thickness deviates from linear relationship with reaction time. (b) Measured [c] of adsorbed R6G vs. h at 25 °C, the symbols indicate the same as in (a).](/image/article/2012/RA/c2ra00754a/c2ra00754a-f4.gif) | ||
| Fig. 4 (a) Measured D of R6G on PNIPAM surfaces vs. h at varied σ = 0.62 (■), 0.33 (●) and 0.13 (▲) chain nm−2 at T = 25 °C. Dashed lines are used to indicate three distinct regions in observed surface dynamical behaviors at σ = 0.62 chain nm−2. Inset: Growth of PNIPAM brush thickness vs. ATRP time at σ = 0.62 (□) and 0.33 (○) chain nm−2. Dashed lines are used to indicate the critical ATRP time, above which the growth of bush thickness deviates from linear relationship with reaction time. (b) Measured [c] of adsorbed R6G vs. h at 25 °C, the symbols indicate the same as in (a). | ||
The measured slow diffusion in region I is contributed to the conformation of PNIPAM layer with low thickness. At h < s, the end-grafted PNIPAM chains collapse and form an actual mushroom surface with resultant high surface heterogeneity; as a result, the surface dynamical behavior of R6G on PNIPAM mushrooms resembles the diffusion on a rough, inhomogeneous surface with high friction and thereby small D. For instance, at σ = 0.62 chain nm−2 or s ≈ 1.4 nm, it is confirmed that PNIPAM brushes of h = 0.8 nm falls into the mushroom regime with an abrupt drop in measured D. On the contrary, at h > 1.4 nm, the end-grafted PNIPAM chains repel each other and extend to the solution to form a polymer brush surface with high surface homogeneity; as a result, the dynamics of R6G on PNIPAM brushes resembles the diffusion on a smooth, homogeneous surface with low friction and thereby large D, which is exhibited in region II of Fig. 4a. However, when h is further increased by extending the ATRP time at constant σ, the chain length of polymer brushes becomes polydispersed, due to the deactivation of partial radicals at the end of the polymer chain during ATRP. The increase in polydispersity of polymer brushes at high h is evident in the observation that the thickness growth of PNIPAM brush deviates from the linear relationship with ATRP time after a certain period, as shown in the insets of Fig. 4a. The decrease in measured D in region III suggests a strong coupling of R6G surface diffusion with the dynamics of PNIPAM brushes of high polydispersity in chain length,33 which is accompanied with a decrease in the segment density predicted in a parabolic distribution in the longitude direction of polymer chains as eqn (7),34–36
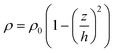 | (7) |
| τ ∼ ρ−3/4 ∼ σ−1/2(1 − (z/h)2)−3/4 | (8) |
According to the scaling prediction in eqn (8) that τs is proportional to σ−1/2, it is suggested that in region II where surface-tethered PNIPAM chains adopt the smooth and uniform brush-like conformation, the surface diffusion of R6G is faster on PNIPAM brushes of higher σ since the coupling degree of R6G thermal activated hopping and PNIPAM chain dynamics can be considered essentially the same with similar R6G–PNIPAM interaction at a constant T. Tentatively, we have plotted the averaged D in region II as a function of σ in Fig. 5. The increase of D as increasing σ is clearly evidenced. For R6G diffusion on PNIPAM brushes, in eqn (5) as:
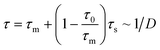
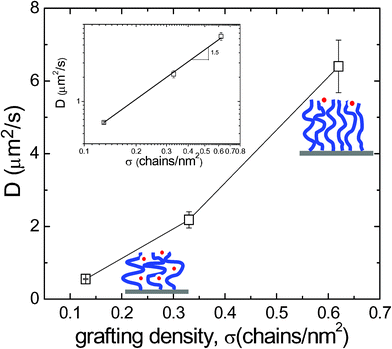 | ||
| Fig. 5 Measured D of R6G on PNIPAM surfaces at T = 25 °C averaged over measurements at varied h in region II as illustrated in Fig. 4a. Inset: the logarithmic plot of D against σ. The fitting slope is about 1.5. | ||
Conclusions
In summary, the surface diffusion of small fluorescent probe molecules, R6G and R123, is investigated with hard, soft and fluid surfaces of distinct dynamic characteristics by FCS at a single molecule level. It is surprising to observe that the surface diffusion of small probe molecules on soft PNIPAM brushes or fluid α-PC lipid bilayer appears slower than on hard OTE monolayer, despite stronger molecular interaction with OTE surface, suggesting the coupling of thermal activated molecular hopping and interfacial dynamics of surface coating films. Therefore, a general and approximate model based on the coupling of thermal activation and the dynamics of underlying surface layer to qualitatively describe molecular diffusion on surfaces of varied dynamic characteristics is proposed. For molecular diffusion on a fluid α-PC lipid bilayer, it is found that the coupling effect is strongly dependent on molecule–surface interaction, where the surface diffusion coefficient of R6G is approximate to the lateral diffusion of lipid molecules yet the surface diffusion coefficient of more weakly adsorbed R123 appears to be much higher. On soft PNIPAM brush surfaces, the coupling effect is further examined with probe diffusion on PNIPAM brushes with varied brush thickness and grafting density. At very low grafting density or very large brush thickness, surface inhomogeneity, which arises from polymer mushroom conformation and low polymer end-segment density, respectively, results in the slowing-down of the interfacial dynamics of PNIPAM chains, and consequently the slowing down of molecular surface diffusion. Furthermore, the surface diffusive dynamics of probe molecules on PNIPAM brushes can be strongly affected by brush grafting density: at high grafting density, R6G molecules diffuse at the brush aqueous interface in a 2-D manner while at low grafting density, R6G could penetrate and diffusion inside the PNIPAM brush layer in a mixed 2-D and 3-D behavior.Acknowledgements
The authors are grateful to the financial support from the US Department of Energy, Office of Basic Sciences, Division of Materials Science and Engineering (Award No. DE-FG02-07ER46390). BJ also acknowledges the financial support from the Center for Sustainable Energy at Notre Dame (cSEND).References
- G. A. Somorjai, Surface Chemistry and Catalysis, Wiley, New York, 1994 Search PubMed.
- M. C. Tringides and Z. Chvoj, Collective Diffusion on Surfaces: Correlation Effects and Adatom Interactions, Kluwer Academic Publishers, Dordrecht, Netherlands, 2001 Search PubMed.
- R. Haberlandt, D. Michel, A. Pöppl and R. Stannarius, Molecules in Interaction with Surfaces and Interfaces, Springer, Berlin, 2004 Search PubMed.
- H. A. Stone, A. D. Stroock and A. Ajdari, Annu. Rev. Fluid Mech., 2004, 36, 381–411 CrossRef.
- G. Brocks, P. J. Kelly and R. Car, Phys. Rev. Lett., 1991, 66, 1729–1732 CrossRef CAS.
- K. Oura, V. G. Lifshits, A. A. Saranin, A. V. Zotov and M. Katayama, Surface Science: An Introduction, Springer, Berlin, 2003 Search PubMed.
- G. Antczak and G. Ehrlich, Surface Diffusion: Metals, Metal Atoms, and Clusters, Cambridge University Press, New York, 2010 Search PubMed.
- M. Q. Zhang, T. Desai and M. Ferrari, Biomaterials, 1998, 19, 953–960 CrossRef CAS.
- D. L. Huber, R. P. Manginell, M. A. Samara, B. I. Kim and B. C. Bunker, Science, 2003, 301, 352–354 CrossRef CAS.
- B. Maier and J. O. Radler, Phys. Rev. Lett., 1999, 82, 1911–1914 CrossRef CAS.
- L. F. Zhang and S. Granick, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9118–9121 CrossRef CAS.
- E. Filippidi, V. Michailidou, B. Loppinet, J. Ruhe and G. Fytas, Langmuir, 2007, 23, 5139–5142 CrossRef CAS.
- H. Tu, L. Hong, S. M. Anthony, P. V. Braun and S. Granick, Langmuir, 2007, 23, 2322–2325 CrossRef CAS.
- M. R. Horton, C. Reich, A. P. Gast, J. O. Radler and B. Nickel, Langmuir, 2007, 23, 6263–6269 CrossRef CAS.
- C. Reznik, Q. Darugar, A. Wheat, T. Fulghum, R. C. Advincula and C. F. Landes, J. Phys. Chem. B, 2008, 112, 10890–10897 CrossRef CAS.
- A. Horner, Y. N. Antonenko and P. Pohl, Biophys. J., 2009, 96, 2689–2695 CrossRef CAS.
- S. Wang and Y. Zhu, Soft Matter, 2010, 6, 4661–4665 RSC.
- C. R. Daniels, C. Reznik, R. Kilmer, M. J. Felipe, M. C. R. Tria, K. Kourentzi, W. H. Chen, R. C. Advincula, R. C. Willson and C. F. Landes, Colloids Surf., B, 2011, 88, 31–38 CrossRef CAS.
- S. Munro, Cell, 2003, 115, 377–388 CrossRef CAS.
- A. Kusumi, C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo and T. Fujiwara, Annu. Rev. Biophys. Biomol. Struct., 2005, 34, 351–354 CrossRef CAS.
- R. C. Advincula, W. J. Brittain, K. C. Caster, J. Rühe, Polymer Brushes: Synthesis, Characterization, Applications, Wiley, New York, 2005 Search PubMed.
- J. Wood and R. Sharma, Langmuir, 1994, 10, 2307–2310 CrossRef CAS.
- S. Wang and Y. Zhu, Langmuir, 2009, 25, 13448–13455 CrossRef CAS.
- B. Jing and Y. Zhu, J. Am. Chem. Soc., 2011, 133, 10983–10989 CrossRef CAS.
- D. Magde, E. Elson and W. W. Webb, Phys. Rev. Lett., 1972, 29, 705–708 CrossRef CAS.
- R. Rigler, U. Mets, J. Widengren and P. Kask, Eur. Biophys. J., 1993, 22, 169–175 CrossRef CAS.
- S. Wang and J. Zhao, J. Chem. Phys., 2007, 126, 091104 CrossRef.
- S. Wang, H. Chang and Y. Zhu, Macromolecules, 2010, 43, 7402–7405 CrossRef CAS.
- V. E. Froude, J. I. Godfroy, S. Wang, H. Dombek and Y. Zhu, J. Phys. Chem. C, 2010, 114, 18880–18885 CAS.
- D. Platikanov, M. Nedyalkov and V. Petkova, Adv. Colloid Interface Sci., 2003, 100–102, 185–203 CrossRef CAS.
- C. E. Heitzman, H. L. Tu and P. V. Braun, J. Phys. Chem. B, 2004, 108, 13764–13770 CrossRef CAS.
- G. Antczak and G. Ehrlich, Surf. Sci. Rep., 2007, 62, 39–61 CrossRef CAS.
- W. Wang, C. Zhang, S. Wang and J. Zhao, Macromolecules, 2007, 40, 9564–9569 CrossRef CAS.
- M. Murat and G. S. Grest, Macromolecules, 1989, 22, 4054–4059 CrossRef CAS.
- P. Y. Lai and K. Binder, J. Chem. Phys., 1991, 95, 9288–9299 CrossRef CAS.
- S. T. Milner, Science, 1991, 251, 905–914 CAS.
- G. E. Yakubov, B. Loppinet, H. Zhang, J. Ruhe, R. Sigel and G. Fytas, Phys. Rev. Lett., 2004, 92, 115501 CrossRef CAS.
- V. N. Michailidou, B. Loppinet, O. Prucker, J. Ruhe and G. Fytas, Macromolecules, 2005, 38, 8960–8962 CrossRef CAS.
- G. Fytas, S. H. Anastasiadis, R. Seghrouchni, D. Vlassopoulos, J. Li, B. J. Factor, W. Theobald and C. Toprakcioglu, Science, 1996, 274, 2041–2044 CrossRef CAS.
- M. Rubinstein and R. H. Colby, Polymer Physics, Oxford University Press, New York, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Lateral diffusive dynamics of α-PC lipid bilayer. See DOI: 10.1039/c2ra00754a |
| This journal is © The Royal Society of Chemistry 2012 |