Capping induced morphology evolution of Rh nanostructures and their electrocatalytic studies
Bhaskar R.
Sathe
*
Department of Chemistry, Dr Babasaheb Ambedkar Marathwada University, Aurangabad, 431 004, India. Fax: +91 240 2400291; Tel: +91 240 2403313E-mail: bhaskarsathe@gmail.com
First published on 7th March 2012
Abstract
A simple and versatile route based on a galvanic displacement approach for the shape-selective evolution of Rh nanostructures followed by their in situ integration assembly on a large scale has been designed and successfully developed by using polyvinylpyrrolidone (PVP) as a shape inducing agent. Comparative studies on the electrocatalytic activity of these morphologies were conducted towards many fuel cell reactions as demonstrated by HCHO oxidation.
1. Introduction
Synthesis of metallic nanostructures followed by their assembly on a desired surface on a large scale, especially using a bottom-up approach, is essential for many of their successful applications in various fields. This has profound implications on understanding their collective behaviour.1 Several recent developments have enabled important applications in electronics, sensing, catalysis and electrochemistry due to their fascinating properties.2 Indeed, different size, shape control and self-organization at nanoscale for metals like Ag, Au, Pd and Pt have been successfully accomplished using a colloidal synthesis approach.3 However, these colloidal nanostructures dispersed in liquids cannot be used directly for many applications, including heterogeneous catalysis and also for device fabrication, because of the challenges in separation, size control, composition and their self-organization and recycling aspects.4 One way to overcome these problems is by using electrochemical tools to precisely control the crystal growth to design high quality nanostructures.5 Consequently, the growth of nanostructures by electrochemical methods has received considerable attention as the presence of an electric field can offer enormous help in controlling even complex morphologies such as fractal geometry and dendritic growth. This method is particularly advantageous because crucial steps such as nucleation and further growth can be regulated by modulating appropriate physical parameters like concentration, number of surface active sites on the template, temperature, pH and electric field. In most of these cases, however, growth occurs under non-equilibrium conditions and interestingly, a dramatic variation in morphology has been observed either at low or high over potentials. For example, potential-dependent morphological evolution for Pt and RuO2 mesostructures has been demonstrated using porous alumina template.6 Despite its high cost, Rh is an attractive material which needs to be exploited properly for fuel cell applications in view of its higher tolerance for CO with respect to Pt and more significantly due to its better size and plane dependant performance.7 Similarly, such a slow evolution of Rh nanostructures under equilibrium conditions could be effected by the simple approach of galvanic exchange, offering a unique way to control the morphology by choosing a suitable redox couple and this approach has been applied successfully to achieve different shapes of various metals and their alloys.8–10 This approach is particularly attractive for preparing nanostructures with well-controlled dimensions and moreover, due to their simplicity, cost effectiveness and high throughput.Accordingly, one way to achieve high quality, one dimensional (1D) nanostructures, is by using solution-phase techniques, via preferential capping mechanisms. It is believed that molecular capping agents play a significant role in the kinetic control of the nanocrystal growth by preferentially adsorbing to specific crystal faces, thus inhibiting growth of those surfaces. Evidence for this selective capping mechanism has been recently demonstrated in the literature by the formation of metal nanowires using PVP as a capping agent.11 One possible explanation is that PVP selectively binds to the {100} facets of Ag while maintaining {111} facets to grow further. However, many questions like morphological evolution, structural modulation and their reactivity correlation, thermodynamic aspects and the growth mechanism have been left unanswered due to the difficulty of size control and involvement of various synthetic parameters. Because of this unusual ability of PVP towards facet selective capping/binding, we have employed it to our microgalvanic approach to modulate Rh ion diffusion towards substrate followed by their reduction. Therefore, this hybrid approach is expected to offer many benefits towards the synthesis of 1D Rh nanostructure.
Herein, we report results of our investigations on such important issues during the synthesis of Rh nanostructures by galvanic displacement approach and focusing more on the role of PVP towards the building of nanorods along with their assemblies on large scale. These measurements allowed sensitive detection of changes in reaction conditions, used to quantitatively assess the growth mechanism for the building of nanostructures and the change in surface properties. Further we have carried out cyclic voltammetric studies to highlight their electrocatalytic reactions at nanostructure interface. Although a detailed mechanism of galvanic displacement is not well understood, an accurate control of key parameters is possible by applying the concepts of corrosion from the mixed potential theory.12 In this case, redox exchange proceeds as long as the oxidized Al3+ ions continue to permeate through the metal film into the solution or until a thick dielectric layer of hydroxide forms, thereby preventing further mass transfer as shown in Scheme 1. Accordingly, Al loses electrons (E°Al3+/Al = −1.66 V vs. SHE) and goes into the solution as solvated Al3+ ions, while Rh3+ ions in the solution accept these electrons (E°Rh3+/Rh = 2.3 V vs. SHE) to form Rh on the surface of Al.
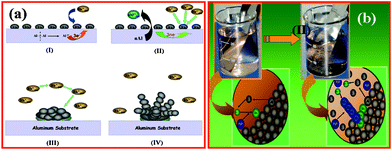 | ||
| Scheme 1 (a) Mechanism proposed to account for the formation of 1D Rh structure on Al substrate as a function of time by using shape inducing agent (PVP). (I) Initial number of active sites on the surface monodisperse nanoparticles, as the reaction proceeds (II–IV) further approach of the ions during growth on the Al surface, (b) Schematic for the formation of 1D Rh structures on Al substrate in the presence of PVP. Initial approach of ions towards lattice Al atoms and growth of Rh-PVP nanostructures on Al surface. | ||
2. Experimental
Synthesis of rhodium nanostructures
For the synthesis of Rh nanostructures; galvanic displacement was carried out by immersing Al foil (thickness 75 μm after etching in 1M NaOH for few minutes to remove the surface oxide layer) in aqueous 1 mM RhCl3. In a typical synthetic procedure, Al foil of bilateral surface area of 2 cm2, was first cleaned by sonication in 50% nitric acid for 2 min. (to remove the native oxide layer from surface) followed by vigorous rinsing with deionized water (∼18 M Ω cm) and finally the residual adsorbed organic impurities were removed by washing with ethanol and drying with nitrogen. This foil was then immersed in 100 mL of aqueous 1 mM RhCl3 solution at room temperature.The effect of capping molecules on the morphological evolution of nanostructured Rh was further demonstrated by a modified galvanic displacement approach, using PVP as the stabilizer. In a typical procedure, similar experiments were carried out using the above procedure in the presence of PVP at appropriate Rh![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PVP ratio (1:5 both solutions in mM). Synthesis of these Rh nanostructures involves electrodeless reduction of Rh ions in the presence of structure modulating PVP molecules using Al as a reducing substrate. The synthetic strategy is briefly (schematically) shown in Scheme 1. The material grown on the surface of aluminum were collected, washed first with 1M NaOH to remove the impurities of alumina embedded into the nanostructures, followed by deionized water and finally with ethanol to remove the impurities like chloride ions and excess RhCl3. This purified material was further characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA). XRD was carried out on a Rigaku, D/MAX IIIB diffractometer using Cu-Kα radiation from a rotating anode X-ray generator operating at 30 kV and 15 mA at room temperature. SEM measurements were carried out with a Leica Stereoscan-440 instrument equipped with a Phoenix energy dispersive analysis of X-ray (EDX) attachment. The XPS measurements were carried out on a VG MicroTech ESCA 3000 instrument at a pressure of > 1 × 10−9 Torr (pass energy of 50 eV, electron takeoff angle 60°, and overall resolution ∼1 eV). The spectra were fitted by using a combined polynomial and Shirley-type background function using “XPS Peak Fitting Program for WIN95/98 XPSPEAK Version 4.1” software. The Shirley-type background fitting function used here having asymmetric Gaussian-Lorentzian sum function. TGA was carried out on TGA Q5000V2.4 TA instruments, build 2–3 thermal analyzer by heating about 2 mg of sample from 50 °C to 900 °C at a rate of 10 °C min−1 in Ar.
:PVP ratio (1:5 both solutions in mM). Synthesis of these Rh nanostructures involves electrodeless reduction of Rh ions in the presence of structure modulating PVP molecules using Al as a reducing substrate. The synthetic strategy is briefly (schematically) shown in Scheme 1. The material grown on the surface of aluminum were collected, washed first with 1M NaOH to remove the impurities of alumina embedded into the nanostructures, followed by deionized water and finally with ethanol to remove the impurities like chloride ions and excess RhCl3. This purified material was further characterized by transmission electron microscopy (TEM), scanning electron microscopy (SEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and thermogravimetric analysis (TGA). XRD was carried out on a Rigaku, D/MAX IIIB diffractometer using Cu-Kα radiation from a rotating anode X-ray generator operating at 30 kV and 15 mA at room temperature. SEM measurements were carried out with a Leica Stereoscan-440 instrument equipped with a Phoenix energy dispersive analysis of X-ray (EDX) attachment. The XPS measurements were carried out on a VG MicroTech ESCA 3000 instrument at a pressure of > 1 × 10−9 Torr (pass energy of 50 eV, electron takeoff angle 60°, and overall resolution ∼1 eV). The spectra were fitted by using a combined polynomial and Shirley-type background function using “XPS Peak Fitting Program for WIN95/98 XPSPEAK Version 4.1” software. The Shirley-type background fitting function used here having asymmetric Gaussian-Lorentzian sum function. TGA was carried out on TGA Q5000V2.4 TA instruments, build 2–3 thermal analyzer by heating about 2 mg of sample from 50 °C to 900 °C at a rate of 10 °C min−1 in Ar.
Electrocatalytic studies
All the electrochemical measurements were performed on an Autolab PGSTAT30 (ECO CHEMIE) instrument using a standard three electrode cell comprising of a Rh nanostructure modified glassy carbon disc as working electrode, Pt foil as the counter electrode and Hg/Hg2SO4 as the reference electrode in 0.5 M H2SO4 at room temperature and the electrochemically active area (ARh) was determined from the adsorption/desorption charge of hydrogen measured from cyclic voltammetry (ca. 230 μC cm−2 for a polycrystalline surface). The glassy carbon electrode (GCE; 3 mm diameter from ECO CHEMIE) was polished before each experiment with 1.0, 0.05 and 0.03 μm alumina slurry and then sonicated in de-ionized water for 10 min. Calculated amount of Rh nanostructures were sonicated in water for 10 min and dropcast on an active tip of the glassy carbon electrode maintaining the same Rh loading on the electrode surface for both. After evaporation under a partial vacuum for 30 min, 10 μL of 0.1 wt% Nafion solution was dropcast on the electrode surface to cover and stabilize the nanostructures. Prior to the analysis, the electrolyte was saturated by passing ultra-high purity nitrogen. Further, in order to measure the electrocatalytic activity of these Rh nanostructures, cyclic voltammetry using a standard three-electrode cell on an Autolab PGSTAT30 (Eco chemie) instrument were carried out. A calculated amount of Rh sample was dropcast on the electrode surface for electrocatalytic studies. Pt foil and a Pt wire were used as the counter and reference electrodes respectively. Formaldehyde (HCHO) oxidation was carried out using a solution of 0.5 M NaOH and 0.5 M HCHO at room temperature; all the voltammograms were measured at a sweep rate of 50 mV s−1. All the potentials here presented with respect to (Hg–HgO and Hg–Hg2SO4) as the reference electrodes respectively.3. Results and discussion
Accordingly, Fig. 1 shows a comparison of SEM images of Rh nanostructures formed on the surface of pretreated Al surface in the presence and absence of capping molecules (PVP) at a reaction time of 5 min. The images clearly depict high coverage of spherical particles of ca. 20 nm size. The particle distribution on the Al surface from both images indicates that some of the initial nanoparticles could act as nucleating centers for further growth. The presence of heterosites on the Al surface (sites where a Rh ad-ion initially had been reduced as Rh (0)) and the limited nucleation density are considered as critical parameters for promoting both the instantaneous growth and the evolution of morphology. Interestingly, it is clear from the SEM images shown in Fig. 1(a) that, the assembly of these spherical nanoparticles into spheres of average diameter of varies as 200 nm. Assuming the shape to be invariant, there is only a minor size variation along with a concomitant interlinking of these nanospheres forming a hierarchical assembly. | ||
| Fig. 1 Scanning electron micrographs of as synthesized Rh (a) nanoflowers and (b) nanorod bundles, in the absence and presence of 1:5 Rh3+:PVP ratio in mM concentration after 5 min respectively. | ||
Moreover, Fig. 1(b) shows the SEM image of Rh nanorods synthesized by the same galvanic displacement approach in the presence of PVP. Interestingly, in the presence of a very small amount of PVP (5 mM), the morphology changes from nanospheres (as shown in Fig. 1a) to bundles of nanorods having 1–1.5 μm length and ∼200 nm diameter. However, each bundle has many nanorods of ∼40 nm diameter. Significantly, this extraordinary 1D growth of the Rh in presence of PVP could be due to its dual function, initially to provide a controlled number of active sites for nucleation on the Al surface and to act as a functional template for the 1D growth of Rh nanostructures.
For example, the role of PVP is to block the Al surface partially by surface adsorption and results in only a small number of active sites being present for galvanic reactions on the surface. Moreover, some of the PVP molecules present in solution act as an additional functional template to selective facet blocking of Rh nanostructures. Initial nucleation of Rh on the Al surface occurs followed by growth of nanostructures at the nucleation sites. The growth of nanostructures is not affected by the crystalline status of the Al substrate. The diffusion of Rh ions towards the limited number of nucleating sites present on the Al surface (not blocked by PVP) is believed to be responsible for the formation of nanorods instead of 3D nanostructures as shown schematically in Scheme 1. Moreover, increasing the amount of PVP the amount of Rh nanostructures on the surface decreases and aggregated types of nanostructures are observed.
Fig. 2 depicts the superimposed XRD patterns of Rh nanostructure especially nanospheres (Fig. 1a) and nanorod bundles (Fig. 1b) with characteristic Rh peaks, indexed to a fcc lattice having (111), (200), (220), (311) and (222) planes.13 A comparison of the relative intensities of the peaks corresponding to Rh-PVP with Rh nanospheres synthesized by the same approach indicates the existence of a morphology/shape dependent preferential growth in different planes. Moreover, these morphology variations along with specific crystallographic facets could be tailored for both selectivity and reactivity towards catalytic reactions.14 For example, the enhanced activity of gold nanoflowers is ascribed to the presence of high index crystal faces such as (220) and (331), which exposes surface irregularities, steps and kinks.15 Similarly, in our case also, Rh nanospheres synthesized by the galvanic displacement approach reveal a comparatively higher intensity of crystal faces like (220) and (331) with respect to nanorods using PVP as a capping agent.
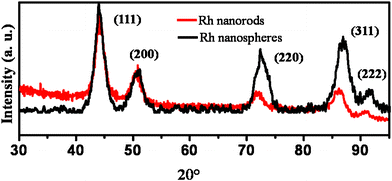 | ||
| Fig. 2 Superimposed X-ray diffraction patterns for Rh nanospheres and Rh nanorods synthesized using the galvanic approach at room temperature. | ||
Interestingly, the growth of nanostructures in principle starts from the lowest energy sites leading to a progressive displacement to the highest energy sites as a function of time, concentration and capping molecules. The rate of growth, initially in (311) plane is more; although there is a decrease inthe case of nanorods (Rh-PVP) indicating the competition between thermodynamic and kinetic factors of the galvanic displacement reaction. For example, typically in the case of nanospheres, the reaction is dominated by non-equilibrium conditions so that growth occurs at the thermodynamically unfavorable high energy (311) plane. This surface induced asymmetry could be the region for the tunable reactivity of the nanostructures. Further, with capping molecules (PVP), these Rh nanospheres become nanorods, via accelerated additional growth along the thermodynamically stable facet direction. A slight variation in the position and intensity with respect to bulk patterns perhaps arises due to the lattice mismatch during kinetically controlled crystal growth.
In order to understand the role of PVP and their local molecular environment involved in the building of nanorods, FTIR studies were conducted after making thin pellets with dry KBr. Accordingly, Fig. 3(a) reveals the superimposed FTIR spectra of Rh nanorods (Rh-PVP) and of only PVP indicating that the capping molecules do interact with Rh. Common peaks at 2805 cm−1 and 1450 cm−1 give information about the orientation of methylene chains along with the symmetric/asymmetric stretching and bending of the C–H of surface bound PVP respectively. Moreover, a strong doublet in the region 1350–1380 cm−1 corresponds to the C–N symmetric and asymmetric stretching16 and these peaks has been shifted to lower side, which could be due to the involvement of a C–N functional group in structure construction. Moreover, the blue shift in the carbonyl frequency from ∼1660 cm−1 to 1700 cm−1, merged into the band corresponding to vinyl group in the region of 1620–1660 cm−1, gives indirect evidence for the involvement of the carbonyl group in the bonding of Rh nanorods.17 The appearance of an additional band at 550 cm−1 in the case of Rh-PVP nanostructures is also attributed to the presence of a Rh–N interaction.18
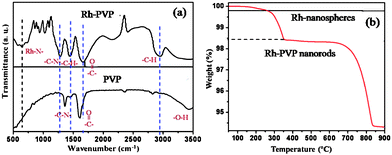 | ||
| Fig. 3 (a) Superimposed FTIR spectra of Rh-PVP (nanorods) along with that for only PVP, (b) Superimposed TG profiles of Rh nanospheres and nanorods performed at a heating rate of 10 °C min−1 in air. | ||
Superimposed thermo-gravimetric profiles of freshly prepared Rh nanospheres (Fig. 1a; without PVP) along with that of Rh nanorods (Fig. 1b; PVP capped) is shown in Fig. 3(b). The profile for Rh nanospheres reveals, at low temperature (below 50 °C) decomposition corresponding to volatile impurities, followed by no weight loss up to 900 °C which could be due to the formation of pure metallic Rh. In comparison, the thermal profile for Rh nanorods shows a three step weight loss. In the first step (150–214 °C) a broad region (weight loss up to 0.15%) is seen attributed to the loss of adsorbed volatile solvent impurities including adsorbed water molecules. In the second step (220–365 °C) a sharp weight loss is seen at 342 °C up to 1.42%, which could be assigned to the thermal desorption of loosely bound PVP molecules. In the third step (360–830 °C) a 4% weight loss could be due to the decomposition of remaining PVP molecules.19 All the results including that of EDS and XPS strongly support this observation and ∼6% of PVP is bound to the surface of Rh nanostructures (not shown here for brevity).
Electrochemical oxidation of organic molecules, including methanol, formaldehyde and formic acid, on Pt, Ru, Rh and their intermetallic alloy electrodes has received great interest due to their importance as anode reactions in fuel cells.20 Although formaldehyde is toxic and not very suitable for commercial fuel cells, its oxidation is important for a complete understanding of the methanol oxidation mechanism because formaldehyde is produced by the partial oxidation of methanol and also because these are linked with CO poisoning on the electrode surface.21 Although expensive, Rh has been extensively investigated as an electrocatalyst for fuel cells and nanostructured Rh in particular has shown several unique features. All the structures are electrocatalytically active for formaldehyde oxidation as the rates at the bare GC electrode and on bulk Rh are much lower than that of the Rh nanostructures. The peak current densities are 0.52, 0.41 and 0.25 mA cm−2 for the reaction on Rh nanospheres, Rh nanorods and bulk Rh as electrocatalysts, respectively (Fig. 4a). In addition to the above observed features, a comparison of the voltammetric response of bulk Rh and nanostructured reveals that the hydrogen adsorption/desorption features of the latter are significantly higher than that of the former. This is presumed to be due to the higher activity of these nanostructures towards hydrogen adsorption/desorption features.
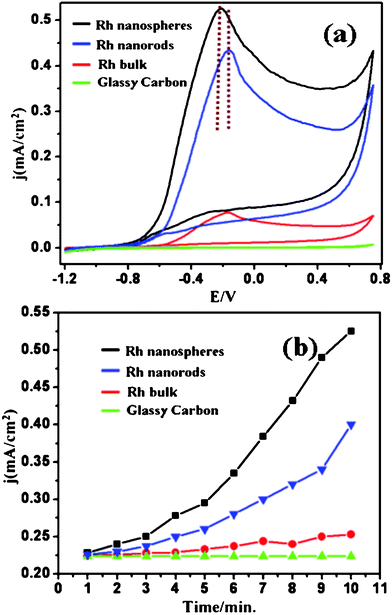 | ||
| Fig. 4 (a) Superimposed cyclic voltammograms in a mixture of 0.5 M NaOH and 0.5 M HCHO solution (first cycle) on Rh nanospheres, nanorods and bulk Rh coated on a glassy carbon working electrode at 50 mV s−1 (b) Respective electrocatalytic activity in terms of variation in current density with time/cycle number (from first 10 cycles) under similar conditions at a peak potential of −0.20 V corresponding of HCHO oxidation. The potentials here presented with respect to Hg–HgO. | ||
More specifically, the electrocatalytic activity of the Rh nanospheres is nearly 2 times higher than that of nanorods synthesised by the same route and both these are significantly higher compared to that of bulk Rh. The improved activity of the Rh nanospheres might be due to their high surface area and preferential crystal facets in high indexing, comparatively low symmetric plane (Fig. 2). Moreover, during the first cycle, only surface atoms are available for electrocatalytic oxidation of formaldehyde and with cycle/time because, of porous nature of nanostructures inner surface/atoms will also participate for electrocatalytic oxidation of formaldehyde and results in an increase in current density with time.22 These results are in good agreement with previously reported results by Rhee and co-workers for Rh, low symmetric plane Rh (100) shows higher electrocatalytic activity than high symmetric Rh (111) for a model electrochemical reaction of perchlorate reduction.23 Hence, from the above observation, we conclude that one of the possible reasons for the varying reactivity of different shapes of Rh nanostructures is due to the difference in the intensity ratio of various crystallographic planes present in the sample, which needs to be verified by carrying out Rietveld refinement. We have also calculated the roughness factor of these electrodes for comparison as shown in Table 1, which clearly indicates that the enhanced electrocatalytic activity of our nanostructures is perhaps not due to the effect of electroactive Rh surface area (ARh). On the other hand, the electrocatalytic results are in good agreement with the intensity ratio of the crystallographic planes, and hence these results indicate the overriding importance of multiple shaped nanostructures in determining the electrocatalytic effect. Table 2 shows a comparison of onset potential and current density of Rh based electrocatalysts with earlier reports on Pt and Pd nanostructures to highlight the importance of the high current density for our Rh nanostructures even though it has a comparatively high onset potential. Furthermore, these Rh nanostructures have an anisotropic morphology with their growth in asymmetric plane that can improve the mass transport for electrocatalytic reactions leading to more effective catalyst utilization.25
| Entry | Rh Electrocatalyst | Electroactive Area (cm2) | Roughness Factor |
|---|---|---|---|
| 1 | Nanospheres | 0.156 | 1.38 |
| 2 | Nanorods | 0.863 | 12.22 |
| 3 | Bulk | 14.38 | 189 |
Conclusions
The present approach for the formation of different morphologies of Rh nanostructures has many intrinsic benefits because of the possibility of tuning the potential changes using molecules like PVP, offering excellent flexibility. In addition, the present approach also helps to generate anisotropic structures by the assistance of controlling the redox properties of the solution and substrate using other passivating molecules. The higher cost and limited availability of Rh, however, pose several limitations for a complete exploitation in fuel cell technology and hence recent efforts have been concentrated on the minimum utilization for similar or enhanced activity. To conclude, we have demonstrated experimental details for the fabrication of plane oriented Rh nanostructures via galvanic displacement using aqueous RhCl3 solution. Further, in order to illustrate the critical importance of controlling some of the major factors affecting their surface reactivity, this approach has been extended towards the effect of PVP molecules on shape selective nanostructure formation of Rh. Promising applications in electrocatalysis have been presented, and other implications of the present studies, for example as fabrication of catalytic nucleation sites for other 1D (nanotubes, nanowires) nanostructure growth, on various substrates are presently under investigation. Future studies are required to focus more on incorporating this method into micro-and nano-lithographic channels to enable site selective catalytic properties. This would ensure more interesting utilization of these nanostructures as important building blocks in complex applications like nanoscale electrical contacts for interfacing a range of different organic and biomolecules, single electron transfer studies, photo-electrochemical and catalytic applications of Rh surfaces. These findings are expected to offer potential benefits in various areas including the design of electrodes for microfuel cells and chemical sensors.Acknowledgements
I would like to thank Dr Vijayamohanan K. Pillai, Director, Central Electrochemical Research Institute (CECRI), Chennai, Karaikudi (India) for his invaluable discussions and guidance, the Council of Scientific and Industrial Research (CSIR), New Delhi (India) for the award of a research fellowship and National Chemical Laboratory Pune (India) for infrastructure are gratefully acknowledged.References
- A. Dong, J. Chen, P. M. Vora, J. M. Kikkawa and C. B. Murray, Nature, 2010, 466, 474 CrossRef CAS.
- P. C. Ray, Chem. Rev., 2010, 110, 5332 CrossRef CAS.
- P. V. Kamat, R. Huehn and R. Nicolaescu, Nanotechnology and the EnvironmentChapter 20 2004, 890, 163. [ACS Symposium Series] Search PubMed.
- K. Ana and T. Hyeon, Nanotoday, 2009, 4, 359 CrossRef.
- D. Pradhan, S. Sindhwani and K.T. Leung, Nanoscale Res. Lett., 2010, 1, 1671 Search PubMed.
- M. Subhramannia, B. K. Balan, B. R. Sathe, I. S. Mulla and V. K. Pillai, J. Phys. Chem. C, 2007, 111, 16593 CAS.
- B. R. Sathe, B. K. Balan and V. K. Pillai, Energy Environ. Sci., 2011, 4, 1029 CAS.
- Z. Peng, H. You, J. Wu and H. Yang, Nano Lett., 2010, 10, 1492 CrossRef CAS.
- B. R. Sathe, B. K. Balan and V. K. Pillai, Appl. Phys. Lett., 2010, 96, 233102 CrossRef.
- B. R. Sathe, B. A. Kakade, A. Kushwaha, M. Aslam and V. K. Pillai, Chem. Commun., 2010, 46, 5671 RSC.
- J. Zhua, C. Kan, H. Li, Y. Cao, X. Ding and J. Wan, J. Cryst. Growth, 2011, 321, 124 CrossRef.
- M. D. Aherne, M. Gara, J. M. Kelly and Y. K. Gunko, Adv. Funct. Mater., 2010, 20, 1329 CrossRef.
- B. R. Sathe, D. S. Shinde and V.K. Pillai, J. Phys. Chem. C, 2009, 113, 9616 CAS.
- Y. Bi and G. Lu, Chem. Commun., 2008, 6402 RSC.
- G. S. Shekhawat and V. P. Dravid, Science, 2005, 310, 89 CrossRef CAS.
- L. M. Harwood and C. J. Moody, Experimental Organic Chemistry: Principles and Practice. Wiley-Blackwell, 1989 ISBN 0632020172 Search PubMed.
- S. Demirci, A. Alaslan and T. Caykara, Appl. Surf. Sci., 2009, 255, 5979 CrossRef CAS.
- S. Pinchas and I. Laulicht, Infrared Spectra of Labeled Compounds, Academic Press: London, 1971; Chapter 9. NIST Vibrational Spectroscopy Database: http://srdata.nist.gov/vb. Search PubMed.
- K. X. Yao and H. C. Zeng, J. Phys. Chem. C, 2007, 111, 13301 CAS.
- A. Miki, S. Ye and M. Osawa, Chem. Commun., 2002, 1500 RSC.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS.
- P. K. Shen, Z. Yan, H. Meng, M. Wu, G. Cui, R. Wang, L. Wang, K. Si and H. Fu, RSC Adv., 2011, 1, 191 RSC.
- C. K. Rhee, M. Wasberg, P. Zelenay and A. Wieckowski, Catal. Lett., 1991, 10, 149 CrossRef CAS.
- M. Subhramannia and V. K. Pillai, J. Mater. Chem., 2008, 18, 5858 RSC.
- G. A. Somorjai, Chemistry in Two Dimensions: Surfaces; Cornell University Press: Ithaca, 1981 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |