DOI:
10.1039/C2RA20130E
(Paper)
RSC Adv., 2012,
2, 3741-3747
Photoinduced electron transfer in supramolecular complexes of a π-extended viologen with porphyrin monomer and dimer†
Received
23rd January 2012
, Accepted 10th February 2012
First published on 13th February 2012
Abstract
A π-extended viologen has been synthesized, forming supramolecular complexes with a freebase tetraphenylporphyrin (H2TPP) and the cofacial porphyrin dimer with an anthracene spacer [H4(DPA)] through π–π interaction in benzonitrile (PhCN). Formation of the H2TPP-BHV2+ supramolecular complex was probed by UV-vis and fluorescence spectra. The fluorescence of H2TPP was strongly quenched by electron transfer from the singlet excited state (1H2TPP*) to BHV2+ in the supramolecular complex. The transient absorption spectrum of the charge-separated (CS) state (H2TPP•+ and BHV•+) was successfully detected by the laser flash photolysis measurements of the H2TPP-BHV2+ supramolecular complex in PhCN. The lifetime of the CS state of the supramolecular complex was determined as 0.37 ms in PhCN at 298 K. The H4(DPA)-BHV2+ supramolecular complex was formed with a larger formation constant as compared to the H2TPP-BHV2+ supramolecular complex because of the stronger π–π interaction of BHV2+ with the cofacial porphyrin dimer and the lifetime of the CS state was determined to be 0.59 ms in PhCN at 298 K.
Introduction
The essential importance of the function of photosynthesis has been well recognized as the energy and environmental problems associated with enormous consumption of fossil fuels evolving unacceptable amounts of CO2 has prompted design and preparation of a large number of donor–acceptor covalently linked ensembles including dyads, triads, tetrads, and pentads that can mimic the electron transfer processes in the photosynthetic reaction center.1–7 However, achieving the long lifetime and the high efficiency of charge separation like natural photosynthesis has often been hampered by the synthetic difficulties of making a number of covalent bonds to connect each component. In contrast to the covalently linked donor–acceptor ensembles, the use of non-covalent binding such as metal–ligand coordination, electrostatic interaction, hydrogen bonds and rotaxane formation has recently merited increasing attention as a simpler but more elegant way to construct electron donor–acceptor ensembles mimicking the biological photosynthetic reaction center model systems.8–20 The development of such supramolecular systems requires molecular modification such as introduction of recognition sites for hydrogen bonding, metal ions for coordination bond formation, and opposite charges for electrostatic interaction. In contrast to these cases, no special molecular modification is required to initiate π–π interactions once appropriate compounds containing π-systems are chosen as components to construct electron donor–acceptor supramolecular ensembles.7–20 However, there has been no report on the introduction of π-conjugation to electron acceptors, which makes possible formation of supramolecular π-complexes with electron donors.
We report herein synthesis of a π-extended viologen, which can form supramolecular complexes with porphyrins through π–π interactions in benzonitrile (PhCN). We have chosen a viologen derivative as an electron acceptor because viologens have frequently been used as an electron mediator for hydrogen production.21–24 Photoexcitation of supramolecular complexes with porphyrin monomers and dimers resulted in efficient photoinduced electron transfer to achieve the long-lived electron-transfer state. Porphyrins and viologen derivatives used in this study are shown in Fig. 1.
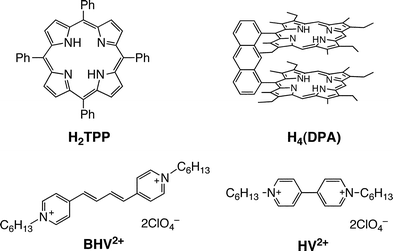 |
| | Fig. 1 Porphyrins and viologens used in this study. | |
Results and discussion
Formation of a supramolecular complex of a π-extended viologen with a porphyrin monomer and dimer
The synthesis and characterization of 1,4-bis(N-hexyl-4-pyridinium)butadiene diperchlorate [BHV2+(ClO4−)2] are described in the experimental section and the electronic supplementary information (ESI) S1 and S2.† First, we examined the supramolecular complex formation of monomer porphyrin (H2TPP) with hexyl viologen (HV2+) and BHV2+. The UV-visible spectrum of H2TPP in PhCN at 298 K was not changed by addition of HV2+ (Fig. 2a). On the contrary, the spectrum of H2TPP was changed upon addition of BHV2+ with appearance of isosbestic points (Fig. 2b). The absorption change at 650 nm is shown in Fig. 3a. Such a saturated dependence on the BHV2+ concentrations indicates that H2TPP forms a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex with BHV2+ (Scheme 1). According to Scheme 1, the absorbance change is given by eqn 1,25 which predicts a linear correlation
:1 complex with BHV2+ (Scheme 1). According to Scheme 1, the absorbance change is given by eqn 1,25 which predicts a linear correlation | | | [H2TPP]0/(A0 − A) = 1/(εc − εp) + (K [BHV2+](εc − εp))−1 | (1) |
between [H2TPP]0/(A0 − A) and [BHV2+]−1, where A0 and A are the absorbance of H2TPP at 650 nm in the absence and presence of BHV2+, and εp and εc are the molar absorption coefficients of H2TPP at 650 nm in the absence and presence of BHV2+, respectively. From a linear plot of [H2TPP]0/(A0 − A) vs. [BHV2+]−1 shown in Fig. 3b, the formation constant (K) of the H2TPP–BHV2+ complex was determined to be 3.1 (±0.5) × 102 M−1 in PhCN at 298 K. In this way, porphyrin formed supramolecular complex with viologen through π–π interactions by using π-extended viologen (BHV2+).
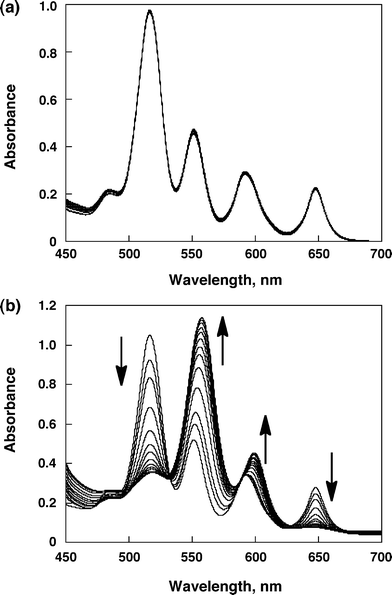 |
| | Fig. 2 (a) UV-visible spectra of H2TPP (6.0 × 10−5 M) in the presence of various concentration of HV2+ (0 to 5.5 × 10−3 M) in PhCN. (b) UV-visible spectra of H2TPP (6.0 × 10−5 M) in the presence of various concentration of BHV2+ (0 to 5.4 × 10−3 M) in PhCN. | |
![(a) Plots of absorbance at 650 nm vs. [BHV2+]. (b) Plots of [H2TPP]0/(A − A0) vs. [BHV2+]−1 at 650 nm.](/image/article/2012/RA/c2ra20130e/c2ra20130e-f3.gif) |
| | Fig. 3 (a) Plots of absorbance at 650 nm vs. [BHV2+]. (b) Plots of [H2TPP]0/(A − A0) vs. [BHV2+]−1 at 650 nm. | |
 |
| | Scheme 1 Supramolecular formation of BHV2+ with H2TPP. | |
The supramolecular complex formation between BHV2+ and cofacial porphyrin dimer (H4(DPA) in Fig. 1)26 was also examined by UV-visible spectroscopy (Fig. S3 in ESI†). An evolution of the spectra was observed in analogy with H2TPP by addition of BHV2+ with isosbestic points (Fig. S4a in ESI†). This indicates that H4(DPA) forms a 1:1 complex with BHV2+ (Scheme 2). The binding constant (K) of this supramolecular complex was determined to be 4.0 (± 0.2) × 102 M−1 from the plot of the absorption change at 500 nm (Fig. S4b in ESI†). This binding constant is larger than for monomer porphyrin, H2TPP, suggesting that the cofacial porphyrin dimer has stronger π–π interactions than the monomer porphyrin, because BHV2+ can be inserted between the two porphyrin rings of H4(DPA).27
 |
| | Scheme 2 Supramolecular formation of BHV2+ with H4(DPA). | |
Photoexcitation of the Q band of H2TPP at 532 nm in PhCN results in fluorescence at λmax = 653 and 714 nm. Addition of BHV2+ to a PhCN solution of H2TPP resulted in a significant change in the fluorescence spectrum of H2TPP (Fig. 4a). The fluorescence intensity of H2TPP decreased to reach a constant value with increasing BHV2+ concentration as the H2TPP–BHV2+ supramolecular complex was formed (inset of Fig. 4a). Virtually the same K value (3.3 (± 0.5) × 102 M−1) was obtained from the change in the fluorescence spectrum of H2TPP in the presence of BHV2+ (Fig. 4b) as the value obtained from the absorption spectral change (vide supra). Such agreement strongly indicates that fluorescence quenching occurs exclusively in the complex rather than intermolecular reaction between the singlet excited state (1H2TPP*) and BHV2+.
![(a) Fluorescence spectra of H2TPP (6.0 × 10−5 M) in the presence of various concentrations of BHV2+ (0 to 4.6 × 10−3 M) in deaerated PhCN. The arrows indicate the direction of change. Inset: Plots of the fluorescence intensity at 720 nm vs. [BHV2+]. (b) Plots of [H2TPP]0/(I − I0) vs. [BHV2+]−1 at 720 nm.](/image/article/2012/RA/c2ra20130e/c2ra20130e-f4.gif) |
| | Fig. 4 (a) Fluorescence spectra of H2TPP (6.0 × 10−5 M) in the presence of various concentrations of BHV2+ (0 to 4.6 × 10−3 M) in deaerated PhCN. The arrows indicate the direction of change. Inset: Plots of the fluorescence intensity at 720 nm vs. [BHV2+]. (b) Plots of [H2TPP]0/(I − I0) vs. [BHV2+]−1 at 720 nm. | |
The fluorescence quenching of H4(DPA) also occurs by BHV2+ in the supramolecular complex between H4(DPA) and BHV2+ (Fig. 4Sa in ESI†). The formation constant (K = 4.0 (± 0.2) × 102 M−1) determined by the fluorescence quenching (Fig. 4b in ESI) agrees with the K value obtained from the absorption spectral change.
One-electron redox potentials of supramolecular complexes
In order to determine the free energy change of photoinduced electron transfer, the redox potentials of H2TPP and BHV2+ were determined by cyclic voltammetry measurements in PhCN (Fig. 5). The one-electron oxidation potential (E0ox) of H2TPP and the reduction potential (E0red) of BHV2+ were determined to be 1.06 V and −0.50 V, respectively (Fig. 5a and 5b). The change in the redox potentials of H2TPP and BHV2+ by the supramolecular complex formation was also examined by CV. The ratio of complexation of the supramolecular complex is calculated as 66% under the experimental conditions, [H2TPP] = 6.0 mM, [BHV2+] = 1.0 mM. The E0ox value of H2TPP was shifted from 1.06 V to 0.84 V upon addition of excess BHV2+ (Fig. 5c). Similarly, the E0red value of BHV2+ was also shifted from −0.50 V to −0.52 V (Fig. 5b and 5d). The energy of the electron-transfer state determined by the E0ox value of H2TPP and the E0red value of BHV2+ is higher than the triplet state of H2TPP (1.43 eV).28 However, the energy of the electron transfer (ET) state in the supramolecular complex determined by the change in the redox potentials by formation of the supramolecular complex is lower than the triplet state of H2TPP. The free energy change of photoinduced electron transfer (ΔG0ET) from 1H2TPP* to BHV2+ in the H2TPP-BHV2+ supramolecular complex in PhCN was determined to be −0.55 eV from the E0ox and E0red values of the H2TPP-BHV2+ supramolecular complex, and the excitation energy (S1 = 1.91 eV)29 of H2TPP in PhCN (Table 1). A similar potential shift was observed in the case of H4(DPA)–BHV2+ (Fig. S5 in ESI†), which is summarized in Table 1. Photoinduced electron transfer in H4(DPA)–BHV2+ is also favorable because of the negative ΔG0ET value (−0.49 eV) as shown in Table 1. Thus, the observed fluorescence quenching of H2TPP and H4(DPA) by BHV2+ results from electron transfer from the singlet excited states (1H2TPP* and 1H4(DPA)*) to BHV2+ in the supramolecular complexes.
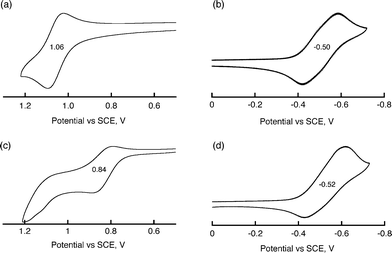 |
| | Fig. 5 Cyclic voltammograms of (a) H2TPP (1.0 × 10−4 M), (b) BHV2+ (1.0 × 10−4 M), (c) H2TPP (1.0 × 10−4 M) with BHV2+ (6.0 × 10−3 M), and (d) H2TPP (6.0 × 10−3 M) with BHV2+ (1.0 × 10−3 M) in deaerated PhCN containing 0.10 M TBAPF6. Scan rate: 100 mV s−1. | |
Table 1 Redox potentials (E0ox and E0red) and driving Force of photoinduced electron transfer (−ΔGET) of supramolecules in PhCN
| Compound |
E
0
ox
vs. SCE (V)a |
E
0
red
vs. SCE (V)a |
ΔGET (eV)a |
|
Values in the parenthesis are given for the uncomplexed form.
|
| H2TPP-BHV2+ |
0.84(1.06) |
−0.52(−0.50) |
−0.55(−0.35) |
| H4(DPA)-BHV2+ |
0.92(1.03) |
−0.56(−0.50) |
−0.49(−0.44) |
Photoinduced electron transfer in supramolecular complexes
The occurrence of the photoinduced electron transfer in the supramolecular π-complex was confirmed by the transient absorption spectra of the π-complex measured in PhCN using nanosecond laser flash photolysis. The transient absorption bands observed in Fig. 6 agree with those of superposition of the absorption bands due to H2TPP•+ and those of BHV•+ in PhCN (Fig. 7).26 Thus, the transient absorption spectrum in Fig. 6 clearly indicates formation of the ET state of H2TPP–BHV2+ by photoinduced electron transfer from the singlet excited state of H2TPP (1H2TPP*) to BHV2+ in the supramolecular complex.
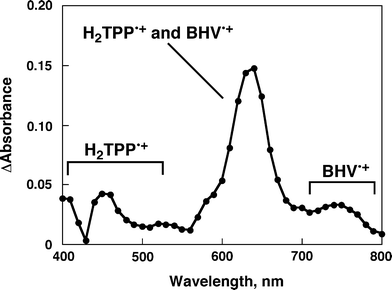 |
| | Fig. 6 Transient absorption spectrum of H2TPP (6.0 × 10−5 M) in the presence of BHV2+ (5.4 × 10−3 M) in deaerated PhCN at 298 K taken at 10 μs after laser excitation at 570 nm. | |
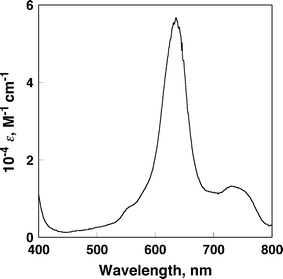 |
| | Fig. 7 Differential spectrum of BHV•+ in the electron-transfer reduction of BHV2+ with tetramethylsemiquinone radical anion in PhCN. | |
In the case of the H4(DPA)–BHV2+ supramolecular complex, similar transient spectra were observed in PhCN (Fig. 8), indicating formation of the ET state of H4(DPA)–BHV2+. Fig. 9a and 9c show time profiles of the decay of the ET states, [H2TPP•+–BHV•+] and [H4(DPA)•+–BHV•+] in PhCN using different laser intensities, respectively. The first-order plots (Fig. 9b and 9d) for the decay time profiles of the ET state at different concentrations afforded linear correlations with the same slope for the long-lived component, irrespective of the difference in concentration of the ET state, whereas the corresponding second-order plots are clearly non-linear and the initial slope varies depending on concentration of the ET state (Fig. S6 in ESI†). The non-linear minor component in the first-order plot (Fig. 9b and 9d) indicates bimolecular charge recombination between charge-separated states or an unbound radical ion pair. Thus, the long-lived decay process is ascribed to back electron transfer from BHV•+ to H2TPP•+ and H4(DPA)•+ in the supramolecular complexes rather than intermolecular back electron transfer.
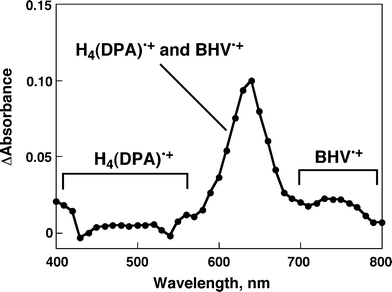 |
| | Fig. 8 Transient absorption spectrum of H4(DPA) (6.0 × 10−5 M) in the presence of BHV2+ (5.5 × 10−3 M) in deaerated PhCN at 298 K taken at 10 μs after laser excitation at 570 nm. | |
![(a) Time profiles of the absorption at 640 nm of H2TPP•+-BHV•+ obtained upon nanosecond flash photolysis (λex = 570 nm) with different laser power in deaerated PhCN at 298 K.; [H2TPP] = 6.0 × 10−5 M; [BHV2+] = 5.4 × 10−3 M. (c) Time profiles of the absorption at 640 nm of H4(DPA)•+-BHV•+ obtained upon nanosecond flash photolysis (λex = 570 nm) with different laser power in deaerated PhCN at 298 K; [H4(DPA)] = 6.0 × 10−5 M; [BHV2+] = 5.5 × 10−3 M. (b, d) First-order plots.](/image/article/2012/RA/c2ra20130e/c2ra20130e-f9.gif) |
| | Fig. 9 (a) Time profiles of the absorption at 640 nm of H2TPP•+-BHV•+ obtained upon nanosecond flash photolysis (λex = 570 nm) with different laser power in deaerated PhCN at 298 K.; [H2TPP] = 6.0 × 10−5 M; [BHV2+] = 5.4 × 10−3 M. (c) Time profiles of the absorption at 640 nm of H4(DPA)•+-BHV•+ obtained upon nanosecond flash photolysis (λex = 570 nm) with different laser power in deaerated PhCN at 298 K; [H4(DPA)] = 6.0 × 10−5 M; [BHV2+] = 5.5 × 10−3 M. (b, d) First-order plots. | |
The lifetimes of the ET state of H2TPP–BHV2+ and H4(DPA)–BHV2+ were determined as 0.37 ms and 0.59 ms, respectively at 298 K. The quantum yields of the ET state of H2TPP-BHV2+ and H4(DPA)–BHV2+ were determined from the comparison of absorbance due to the triplet excited state of H2TPP (ε430 = 83000 M−1 cm−1)30 and the absorption of the CS state (ε640 = 54000 M−1 cm−1)31 to be 0.84 and 0.83, respectively. The ε640 value was determined from the absorption spectra of BHV•+, which was produced by the electron transfer reduction of BHV2+ with tetramethylsemiquinone radical anion in PhCN. (Fig. 7). Tetramethylsemiquinone radical anion was produced by the proportionation reaction of tetramethyl-p-benzoquinone and tetramethylhydroquinone in the presence of a base.28
Formation of the ET state was further confirmed by the EPR measurements after photoirradiation of H4(DPA)–BHV2+ in frozen PhCN at 173 K. The resulting EPR spectrum is shown in Fig. 10a.32 The observed isotropic EPR signal at g = 2.0028 consists of the superposition of that of H4(DPA)•+ (isotropic signal at g = 2.0029 in Fig. 10b) produced by the one-electron oxidation of H4(DPA) with [Fe(bpy)3]3+ (bpy = 2,2′-bipyridine) and that of BHV•+ (isotropic signal at g = 2.0027) produced by the one-electron reduction of BHV2+ with a tetramethylsemiquinone radical anion (Fig. 10c).33
3 (8.6 × 10−5 M). (c) EPR spectrum of a MeCN solution of BHV•+ at 173 K generated by the electron-transfer reduction of BHV2+ (2.9 × 10−3 M) with tetramethylsemiquinone radical anion (9.8 × 10−5 M). The asterisk * denotes Mn2+ marker.](/image/article/2012/RA/c2ra20130e/c2ra20130e-f10.gif) |
| | Fig. 10 (a) EPR spectrum of (a) charge-separated state obtained from photoirradiation of a deaerated PhCN solution containing H4(DPA) (6.0 × 10−5 M) and BHV2+ (5.5 × 10−3 M) with a high pressure Hg lamp at 173 K. (b) EPR spectrum of a MeCN solution of H4(DPA)•+ at 173 K generated by the electron transfer oxidation of H4(DPA) (1.5 × 10−4 M) with [Fe(bpy)3](PF6)3 (8.6 × 10−5 M). (c) EPR spectrum of a MeCN solution of BHV•+ at 173 K generated by the electron-transfer reduction of BHV2+ (2.9 × 10−3 M) with tetramethylsemiquinone radical anion (9.8 × 10−5 M). The asterisk * denotes Mn2+ marker. | |
Conclusion
In conclusion, a π-extended viologen (BHV2+) forms supramolecular π-complexes with a porphyrin monomer and dimer, which affords long-lived ET states upon the photoexcitation. Such π-extension of electron acceptors provides a new strategy for formation of supramolecular π-complexes to achieve efficient photoinduced electron transfer and slow back electron transfer.
Experimental
General
1H NMR spectra were measured on a JEOL JMN-AL300 (300 MHz). Matrix-assisted laser desorption/ionization (MALDI) time-of-flight mass spectra (TOF) were measured on a Kratos Compact MALDI I (Shimadzu).
Materials
A cofacial bisporphyrin, H4(DPA) [DPA = 1,8-bis[5-(2,8,13,17-tetraethyl-3,7,12,18-tetramethylporphyrinyl)]anthracene] was synthesized according to literature.26 5,10,15,20-Tetraphenyl-21H,23H-porphyrin (H2TPP) and 4-(bromomethyl)pyridine hydrobromide was obtained from Aldrich Chemical Co., Inc. Tetra-n-butylammonium perchlorate (TBAP) used as a supporting electrolyte for electrochemical measurements was obtained from Tokyo Chemical Industry (TCI). Triphenylphosphine, β-(4-pyridyl)acrolein oxalate, 1-bromohexane, sodium carbonate, sodium hydroxide, dichloromethane, methanol, ethanol, acetone, benzonitrile (PhCN) and acetonitrile (MeCN) were purchased from Wako Pure Chemical Ind., Ltd. Benzene was obtained from Nacalai Tesque, Inc. PhCN and MeCN were purified by successive distillation over calcium hydride.34
Synthesis of 1,4-bis(N-hexyl-4-pyridinium)butadiene diperchlorate [BHV2+(ClO4−)2]
A solution of 4-(bromomethyl)pyridine hydrobromide (1.8 g, 7 mmol) in H2O was basified with Na2CO3 and extracted with benzene. To this organic layer was added PPh3 (7.9 g, 30 mmol) and stirred at room temperature for 60 h under air. The reaction mixture was then precipitated out of benzene, filtered to give the cream colored solid (2.1 g, 4.8 mmol, 69% yield). To the solution of that solid (0.87 g, 2 mmol) and β-(4-pyridyl)acrolein oxalate (0.45 g, 2 mmol) in CH2Cl2 were added a 50% NaOH solution under nitrogen atmosphere. The reaction mixture was stirred for 2 h at room temperature, and then diluted with CH2Cl2 and H2O. The organic layer was concentrated in vacuo, and the residue was dissolved in anhydrous EtOH. Passing HCl gas through the EtOH solution gave a brown precipitate, which was collected and then dissolved in H2O. The solution was basified with Na2CO3 and CH2Cl2 extraction followed by solvent evaporation gave 1,4-di(4-pyridyl)butadiene. The product was recrystallized from MeOH–H2O (1:1) to give a brown solid (75 mg, 0.36 mmol, 18% yield). 1,4-Di(4-pyridyl)butadiene (75 mg, 0.36 mmol) and 1-bromohexane (206 mg, 1.26 mmol) were dissolved in MeCN. The mixture was heated to reflux for 72 h. After cooling, the brown precipitate was filtered and rinsed with Et2O. The obtained dibromide (186 mg, 0.35 mmol) and sodium perchlorate (2.3 g, 18.7 mmol) were dissolved H2O and stirred for 20 h. The yellow precipitate was rinsed with EtOAc and recrystallized from acetone and yellow solid (172 mg, 0.3 mmol) was obtained.351H NMR (300 MHz, CD3CN) δ 8.55 (d, J = 6.6 Hz, 4H), 8.03 (d, J = 6.6 Hz, 4H), 7.64 (dd, J = 3.0 Hz and 9.3 Hz, 2H), 7.14 (dd, J = 3.0 Hz, 9.3 Hz, 2H), 4.44 (t, J = 7.2 Hz, 4H), 1.31–1.50 (m, 4H), 1.34 (m, 12H), 0.90 (t, J = 6.6 Hz, 6H); MALDI-TOF MS m/z 378.31 (BHV•+ requires 378.30). Anal. Calcd for C26H38Cl2N2O8: C, 54.07; H, 6.63; N, 4.85. Found: C, 53.78; H, 6.42; N, 4.80.
Photophysical measurements
UV-visible absorption spectra were obtained on a Shimadzu UV-3100PC spectrometer or a Hewlett Packard 8453 diode array spectrophotometer at 298 K. Corrected fluorescence spectra were taken using a SHIMADZU spectrofluorophotometer (RF-5300PC). The solutions were deaerated by argon purging for 15 min prior to the measurements. For nanosecond laser flash photolysis experiments, deaerated PhCN solutions were excited by a Panther OPO pumped by Nd:YAG laser (Continuum, SLII-10, 4–6 ns fwhm) at λ = 570 nm. The photochemical reactions were monitored by continuous exposure to a Xe-lamp (150 W) as a probe light and a photomultiplier tube (Hamamatsu 2949) as a detector. Transient absorption spectra were recorded using fresh solutions in each laser excitation.
Electrochemical measurements
Cyclic voltammetry measurements were performed at 298 K on an ALS-630B voltammetric analyzer in deaerated solvent containing 0.10 M TBAPF6 as a supporting electrolyte at 298 K. A conventional three-electrode cell was used with a platinum working electrode and a platinum wire as a counter electrode. The measured potentials were recorded with respect to the Ag/AgNO3 (1.0 × 10−2 M). The E0ox and E0red values (vs. Ag/Ag+) are converted to those vs. SCE by adding 0.29 V, respectively.36
ESR measurements
An argon-saturated PhCN solution of H4(DPA) (6.0 × 10−5 M) and BHV2+ (5.5 × 10−3 M) was irradiated at 173 K with a high pressure mercury lamp (USH-1005D) through a water filter focused at the sample cell in the ESR cavity. The ESR spectra were taken on a JEOL JES-RE1XE and were recorded under non-saturating microwave power conditions. The magnitude of the modulation was chosen to optimize the resolution and the signal to noise ratio (S/N) of the observed spectra. The g values were calibrated using an Mn2+ marker.
Acknowledgements
This work was financially supported by a Grant-in-Aid (No. 20108010 and 23750014) and the Global COE (center of excellence) program “Global Education and Research Center for Bio-Environmental Chemistry” of Osaka University from MEXT, Japan, NRF/MEST of Korea through WCU (R31-2008-000-10010-0) and GRL (2010-00353) programs, and The Centre National de la Recherche Scientifique (ICMUB, UMR 6302), France.
References
-
(a) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2001, 34, 40 CrossRef CAS;
(b) D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890 CrossRef CAS.
-
(a) M. R. Wasielewski, Chem. Rev., 1992, 92, 435 CrossRef CAS;
(b) M. R. Wasielewski, Acc. Chem. Res., 2009, 42, 1910 CrossRef CAS.
-
(a) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 676 CrossRef;
(b) N. Martin, L. Sanchez, M. A. Herranz, B. Illescas and D. M. Guldi, Acc. Chem. Res., 2007, 40, 1015 CrossRef CAS.
-
(a) S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609 RSC;
(b) S. Fukuzumi, Bull. Chem. Soc. Jpn., 2006, 79, 177 CrossRef CAS;
(c) S. Fukuzumi, Phys. Chem. Chem. Phys., 2008, 10, 2283 RSC;
(d) K. Ohkubo and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2008, 12, 993 CrossRef CAS;
(e) S. Fukuzumi and T. Kojima, J. Mater. Chem., 2008, 18, 1427 RSC.
-
(a) S. Fukuzumi, Eur. J. Inorg. Chem., 2008, 1351 CrossRef CAS;
(b) S. Fukuzumi, Y. Yamada, T. Suenobu, K. Ohkubo and H. Kotani, Energy Environ. Sci., 2011, 4, 2754 RSC.
-
(a) H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607 CrossRef CAS;
(b) H. Imahori, D. M. Guldi, K. Tamaki, Y. Yoshida, C. Luo, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 6617 CrossRef CAS;
(c) D. M. Guldi, H. Imahori, K. Tamaki, Y. Kashiwagi, H. Yamada, Y. Sakata and S. Fukuzumi, J. Phys. Chem. A, 2004, 108, 541 CrossRef CAS.
-
(a) D. Curiel, K. Ohkubo, J. R. Reimers, S. Fukuzumi and M. J. Crossley, Phys. Chem. Chem. Phys., 2007, 9, 5260 RSC;
(b) H. Imahori, Y. Sekiguchi, Y. Kashiwagi, T. Sato, Y. Araki, O. Ito, H. Yamada and S. Fukuzumi, Chem.–Eur. J., 2004, 10, 3184 CrossRef CAS;
(c) S. H. Lee, A. G. Larsen, K. Ohkubo, J. R. Reimers, S. Fukuzumi and M. J. Crossley, Chem. Sci., 2012, 3, 257 RSC;
(d) C. P. Gros, F. Brisach, A. Meristoudi, E. Espinosa, R. Guilard and P. D. Harvey, Inorg. Chem., 2007, 46, 125 CrossRef CAS;
(e) M. A. Filatov, R. Guilard and P. D. Harvey, Org. Lett., 2010, 12, 196 CrossRef CAS.
-
(a)
D. Gust, T. A. Moore and A. L. Moore, In From Non-Covalent Assemblies to Molecular Machines, ed. J.-P. Sauvage and P. Gaspard (ed.), Wiley-VCH, Weinheim, 2011, pp. 321–354 Search PubMed;
(b)
V. Balzani, A. Credi and M. Venturi, in Organic Nanostructures, ed. J. L. Atwood and J. W. Steed, Wiley-VCH, Weinheim, 2008, pp. 1–31 Search PubMed.
-
(a) L. Martin-Gomis, K. Ohkubo, F. Fernandez-Lazaro, S. Fukuzumi and Á. Sastre-Santos, Chem. Commun., 2010, 46, 3944 RSC;
(b) M. E. El-Khouly, K.-J. Han, K.-Y. Kay and S. Fukuzumi, ChemPhysChem, 2010, 11, 1726–1734 CAS.
-
(a) D. I. Schuster, K. Li, D. M. Guldi, A. Palkar, L. Echegoyen, C. Stanisky, R. J. Cross, M. Niemi, N. V. Tkachenko and H. Lemmetyinen, J. Am. Chem. Soc., 2007, 129, 15973 CrossRef CAS;
(b) T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Phys. Chem. C, 2010, 114, 14290 CrossRef CAS;
(c) F. J. Cespedes-Guirao, K. Ohkubo, S. Fukuzumi, Á. Sastre-Santos and F. Fernandez-Lazaro, J. Org. Chem., 2009, 74, 5871 CrossRef CAS.
-
(a) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC;
(b) R. Chitta and F. D'Souza, J. Mater. Chem., 2008, 18, 1440 RSC;
(c) F. D'Souza and O. Ito, Coord. Chem. Rev., 2005, 249, 1410 CrossRef CAS.
-
(a) J. L. Sessler, E. Karnas, S. K. Kim, Z. Ou, M. Zhang, K. M. Kadish, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 15256 CrossRef CAS;
(b) S. Fukuzumi, K. Ohkubo, Y. Kawashima, D. Kim, J. S. Park, A. Jana, V. M. Lynch, D. Kim and J. L. Sessler, J. Am. Chem. Soc., 2011, 133, 15938 CrossRef CAS.
-
(a) T. Kojima, T. Honda, K. Ohkubo, M. Shiro, T. Kusukawa, T. Fukuda, N. Kobayashi and S. Fukuzumi, Angew. Chem., Int. Ed., 2008, 47, 6712 CrossRef CAS;
(b) T. Honda, T. Nakanishi, K. Ohkubo, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 10155 CrossRef CAS;
(c) J. Rosenthal, J. M. Hodgkiss, E. R. Young and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 10474 CrossRef CAS.
- S. Fukuzumi, T. Honda, K. Ohkubo and T. Kojima, Dalton Trans., 2009, 3880 RSC.
- M. Tanaka, K. Ohkubo, C. P. Gros, R. Guilard and S. Fukuzumi, J. Am. Chem. Soc., 2006, 128, 14625 CrossRef CAS.
-
(a) A. Takai, M. Chkounda, A. Eggenspiller, C. P. Gros, M. Lachkar, J.-M. Barbe and S. Fukuzumi, J. Am. Chem. Soc., 2010, 132, 4477 CrossRef CAS;
(b) A. Takai, C. P. Gros, J.-M. Barbe and S. Fukuzumi, Phys. Chem. Chem. Phys., 2010, 12, 12160 RSC.
-
(a) M. E. El-Khouly, D. K. Ju, K.-Y. Kay, F. D'Souza and S. Fukuzumi, Chem.–Eur. J., 2010, 16, 6193 CrossRef CAS;
(b) F. D'Souza, C. A. Wijesinghe, M. E. El-Khouly, J. Hudson, M. Niemi, H. Lemmetyinen, N. V. Tkachenko, M. E. Zandler and S. Fukuzumi, Phys. Chem. Chem. Phys., 2011, 13, 18168 RSC.
-
(a) S. Fukuzumi, K. Saito, K. Ohkubo, V. Troiani, H. Qiu, S. Gadde, F. D'Souza and N. Solladie, Phys.
Chem. Chem. Phys., 2011, 13, 17019 CAS;
(b) S. Fukuzumi, K. Saito, K. Ohkubo, T. Khoury, Y. Kashiwagi, M. A. Absalom, S. Gadde, F. D'Souza, Y. Araki, O. Ito and M. J. Crossley, Chem. Commun., 2011, 47, 7980 RSC;
(c) S. Fukuzumi and Y. Kashiwagi, J. Porphyrins Phthalocyanines, 2007, 11, 368 CrossRef CAS.
-
(a) H. Nobukuni, Y. Shimazaki, H. Uno, Y. Naruta, K. Ohkubo, T. Kojima, S. Fukuzumi, S. Seki, H. Sakai, T. Hasobe and F. Tani, Chem.–Eur. J., 2010, 16, 11611 CrossRef CAS;
(b) M. Murakami, K. Ohkubo, T. Hasobe, V. Sgobba, D. M. Guldi, F. Wessendorf, A. Hirsch and S. Fukuzumi, J. Mater. Chem., 2010, 20, 1457 RSC.
-
(a) F. D'Souza, N. K. Subbaiyan, Y. Xie, J. P. Hill, K. Ariga, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 16138 CrossRef CAS;
(b) F. D'Souza, E. Maligaspe, K. Ohkubo, M. E. Zandler, N. K. Subbaiyan and S. Fukuzumi, J. Am. Chem. Soc., 2009, 131, 8787 CrossRef CAS;
(c) T. Kojima, T. Nakanishi, H. Honda and S. Fukuzumi, J. Porphyrins Phthalocyanines, 2009, 13, 14 CrossRef CAS;
(d) T. Kojima, T. Nakanishi, R. Harada, K. Ohkubo, S. Yamauchi and S. Fukuzumi, Chem.–Eur. J., 2007, 13, 8714 CrossRef CAS.
- T. S. Teets and D. G. Nocera, Chem. Commun., 2011, 47, 9268 RSC.
-
(a) H. Kotani, T. Ono, K. Ohkubo and S. Fukuzumi, Phys. Chem. Chem. Phys., 2007, 9, 1487 RSC;
(b) H. Kotani, K. Ohkubo, Y. Takai and S. Fukuzumi, J. Phys. Chem. B, 2006, 110, 24047 CrossRef CAS.
- M. Ogawa, G. Ajayakumar, S. Masaoka, H.-B. Kraatz and K. Sakai, Chem.–Eur. J., 2011, 17, 1148 CrossRef CAS.
- P. Du, J. Schneider, P. Jarosz and R. Eisenberg, J. Am. Chem. Soc., 2006, 128, 7726 CrossRef CAS.
- S. Fukuzumi, Y. Kondo, S. Mochizuki and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1989, 1753 RSC.
- S. Faure, C. Stern, R. Guilard and P. D. Harvey, J. Am. Chem. Soc., 2004, 126, 1253 CrossRef CAS.
- A similar sandwiched π-complex between H4(DPA) and acridinium cation was reported in ref.15.
-
(a) A. Harriman, J. Chem. Soc., Faraday Trans. 2, 1979, 75, 1532 RSC;
(b) E. G. Ermolina, R. T. Kuznetsova, R. M. Gadirov, G. V. Maier, N. N. Semenishin, N. V. Rusakova and Y. V. Korovin, High Energy Chem., 2010, 44, 387 CrossRef CAS.
- The energies of the 0-0 transition between the S1 and the S0 state were determined by averaging the energies of the corresponding (0,0) peaks in the fluorescence and the absorption bands..
-
(a) L. Pekkarinen and H. Linschitz, J. Am. Chem. Soc., 1960, 82, 2407 CrossRef CAS;
(b) H. Gratz and A. Penzkofer, Chem. Phys., 2000, 254, 363 CrossRef CAS.
-
(a) C. Inisan, J.-Y. Saillard, R. Guilard, A. Tabard and Y. Le Mest, New J. Chem., 1998, 22, 823 RSC;
(b) K. Okamoto, K. Ohkubo, K. M. Kadish and S. Fukuzumi, J. Phys. Chem. A, 2004, 108, 10405 CrossRef CAS.
- We could not observe the signal with zero-field splitting pattern around g = 2 (3230 G ± 300 G) and the triplet marker signal at g = 4 due to the triplet charge-separated state in this study. The spin–spin interaction of radical ion pair may not be strong..
- K. Ohkubo, K. Suga, K. Morikawa and S. Fukuzumi, J. Am. Chem. Soc., 2003, 125, 12850 CrossRef CAS.
-
W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, 5th ed., Butterworth Heinemann. Amsterdam, 2003 Search PubMed.
- S. Fukuzumi, Jpn. Kokai Tokkyo Koho, JP, 2005, 2005255603 Search PubMed.
-
C. K. Mann and K. K. Barnes, Electrochemical Reactions in Nonaqueous Systems, Marcel Dekker, New York, 1990 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.