Efficient electrocatalytic oxidation of water: minimization of catalyst loading by an electrostatic assembly of hydrous iridium oxide colloids†
Min-Chieh
Chuang
and
Ja-an Annie
Ho
*
Department of Biochemical Science and Technology, National Taiwan University, Taipei 10617, Taiwan. E-mail: jaho@ntu.edu.tw; Fax: +886 2 3366 2271; Tel: +886 2 3366 4438; +886 2 3366 4439
First published on 16th March 2012
Abstract
Exploiting an effective catalyst has been an essential and significant task in the development of electrochemical water oxidation. A highly electrocatalytic iridium oxide colloidal electrode, prepared by the electrostatic assembly fashion, is reported herein. Poly(allylamine hydrochloride) (PAH) was utilized to facilitate the formation of stable PAH/iridium oxide colloid clusters, resulting in a high catalytic utility (turnover efficiency of 6.36 ± 0.25 s−1 at low overpotential, η = 0.28 V) towards the oxygen evolution reaction (OER), with the smallest Ir loading (Γcov = 2.3 × 10−9 mol cm−2) of any yet reported. A variety of examinations and approaches were conducted to gain better understanding of such electrostatically assembled architecture and to identify hexahydroxyiridate(IV), [Ir(OH)6]2−, as the key active catalytic species.
1. Introduction
Increasing attention has been paid to explore clean and safe alternative energy sources to solve the recent problems relating to energy and the environment.1 Artificial photosynthesis is deemed to be one of the most promising energy-providing systems, in which the production of oxygen (O2) and hydrogen (H2) is implemented by harnessing photo-electrochemical or electrochemical splitting of water.2In order to promote the splitting of water, it is necessary to direct a more efficient electrochemical oxidation of water to O2 (eqn (1)), owing to its slow electrode kinetics,3 thus necessitates a search for an efficient catalyst to function at high turnover rates and low overpotentials.
| 2H2O → O2 + 4H+ + 4e− | (1) |
Enormous efforts have been devoted to developing such a catalyst4–6 for the oxygen evolution reaction (OER). Certain metal complexes, such as manganese, ruthenium, and iridium complexes, were proclaimed to be efficient catalysts for this purpose.7–11 Of these metal complexes, iridium oxide (IrOx) was extensively studied as a promising candidate, identified by its comparative activity and higher stability over the RuO2, a well-studied heterogeneous catalyst in both electrochemical and photochemical systems. Vapor-phase techniques such as pulsed-laser deposition and chemical vapor deposition,12–14 as well as anodic deposition15–17, have been performed to prepare nano-structured IrO2 thin films in numerous studies. However OER is still recognized as a potential bottleneck as a result of the low fraction of the electroactive Ir site (Γea), which hinders the promotion of turnover frequency (TOF) and reduction of overpotential (η).
This article describes a novel approach yielding an electrostatically-assembled iridium oxide colloidal catalytic electrode from an iridium oxide nanoparticle solution. Nanoparticles (NPs) have been of interest in many fields because of their large surface-to-volume ratio and size-dependent properties. Recently, high-surface-area films of iridium oxide have been made from colloidal suspensions by means of electro-flocculation and self-assembly fashion. Murray and coworkers18 described a formation of stable, adherent, mesoporous film made of 2 nm diameter IrOx nanoparticles on glassy carbon electrode (GCE), by a controlled potential electro-flocculation from alkaline colloidal solutions (pH 13). The authors proposed an IrOx formation mechanism in which the protons released by water oxidation lowered the local pH at the electrode surface to partly neutralize the hydroxyl capping layer that had served to stabilize the IrOx film. Subsequent studies agreed with such a mechanism to anodically generate further stable hydrous iridium oxide (IrOx·nH2O) thin film, in either acidic (pH 1)19 or neutral pH conditions,20 towards OER at lower overpotential and higher turnover frequency. While electrical power is required to exert such electro-flocculation process, a deposition implemented by a spontaneous self-assembly is relatively desirable in order to fit the eco-friendly protocol with minimal energy consumption. Yagi and coworker21,22 reported a 50–100 nm diameter citrate-stabilized IrO2 nanoparticle which was assembled on ITO electrodes to result in an average TOF of ∼6.6 s−1. However η is higher than 0.5 V (at pH 7). Interestingly, the electroactive Ir loading in these previous studies ranged from 10−7∼10−8 mol cm−2, which is considered as one of key factors hampering the large scale applications since iridium is the least abundant metal in the earth.
In order to address the challenges, we aim to form a stable and highly catalytic IrOx·nH2O electrode at minimal Ir loading towards an efficient OER, in which a catalyst architecture efficiently producing catalytic Ir sites is expectantly achieved. GCE exhibited IrIV/IrIII redox waves in the IrOx·nH2O colloidal (1–2 nm) solution, yet such the characteristic waves faded as an increase of sweep cycles in the cyclic voltammogram and the time exposed to a blank buffer. This phenomenon appeared to be a behaviour in connection to an adsorption of the hydrous iridium oxide colloid onto GCE and the possible desorption in the colloid-deficient solution. In this study we utilized a self-assembly of polyallylamine hydrochloride (PAH), a positively charged polymer, to substantially mitigate the desorption of the adsorbed iridium oxide colloid, prompted the quantity of the electroactive Ir sites, and enhanced the IrIV/IrIII redox reversibility (Fig. S1, ESI†). PAH was assembled on the GCE surface to subsequently deposit the iridium oxide colloids electrostatically. To our best knowledge, this is the first electrostatic assembly of ligand-free iridium oxide colloid used to enhance the efficiency of OER, in addition to the characterization of the resulting active catalyst. A series of experiments were, furthermore, conducted to gain better understanding of catalytic property and to identify the active catalytic species.
2. Experimental
2.1 Chemicals and materials
Potassium hexachloroiridate(IV) (K2IrCl6), potassium phosphate monobasic (KH2PO4, ≥99.0%), potassium phosphate dibasic (K2HPO4, ≥99.0%), sodium chloride (NaCl, ≥99.5%), polyallylamine hydrochloride (PAH, Mw ∽15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), polyvinyl alcohol (PVA, Mw 30000–70000), polyacrylic acid, sodium salt (PAA, Mw∼1200) were obtained from Sigma-Aldrich Corp. (St. Louis, MO) and used as supplied. Sodium hydroxide (NaOH), nitric acid (HNO3), sulfuric acid (H2SO4), and hydrochloric acid (HCl) were purchased from Riedel-de Haën. Deionized water (>18 MΩ cm) from a Direct-Q system (Millipore, Billerica, MA) was used to prepare all aqueous solutions.
000), polyvinyl alcohol (PVA, Mw 30000–70000), polyacrylic acid, sodium salt (PAA, Mw∼1200) were obtained from Sigma-Aldrich Corp. (St. Louis, MO) and used as supplied. Sodium hydroxide (NaOH), nitric acid (HNO3), sulfuric acid (H2SO4), and hydrochloric acid (HCl) were purchased from Riedel-de Haën. Deionized water (>18 MΩ cm) from a Direct-Q system (Millipore, Billerica, MA) was used to prepare all aqueous solutions.
2.2 Preparation of colloid and electrodes
IrOx colloids were prepared from 2.5 mM aqueous K2IrCl6 adjusted to pH 13 with 6.25 M NaOH to obtain a yellow solution, which was then heated at 90 °C for 20 min, and immediately cooled in an ice bath. The resulting cold blue solution was adjusted to pH 1 by adding 3 M HNO3 and was stirred continuously for 80 min until the solution turned deep blue. The solution was stored at 4 °C until use.Prior to functionalization of glassy carbon electrode (GCE), the electrode was polished with 0.05 μm alumina powder suspension (in water) and thoroughly rinsed with distilled water. An aliquot (25 μL) of 6.25 M aqueous NaOH was loaded onto the electrode surface and kept at 25 °C for 20 min. The surface of the electrode was then dried with nitrogen after removing the NaOH. Subsequently, 25 μL PAH solution (5 mg mL−1 in 0.05 M NaCl) was dropped onto the electrode surface and left to react for 20 min. The resulting electrode was rinsed with 0.05 M NaCl to produce a PAH-functionalized GCE. Concurrently, an IrOx colloidal assembly solution was prepared by diluting 60 μL of the IrOx colloid (pH 1) in an 800 μL phosphate buffer (0.05 M, pH 7.2). The PAH-functionalized GCE was then immersed in the assembly solution for the desired period to obtain the electrodes used in OER investigation. To prepare assembly solutions with varied pH, the IrOx colloidal assembly solution was titrated with the appropriate quantity of aqueous NaOH or HNO3.
2.3 Measurements and characterizations
A CH Instruments model 633C Electrochemical Analyzer (Austin, TX) was used in all electrochemical measurements performed under ambient atmosphere. Cyclic voltammetric characterization of the assembled electrode was conducted, which was held in a conventional single-compartment three-electrode electrochemical cell equipped with an Ag/AgCl reference electrode (CHI111, CH Instruments) and a 0.5 mm diameter platinum wire counter electrode at 50 mV s−1 in the following solutions: pH 7.2 phosphate buffer (0.05 M), pH 1.2 H2SO4 solution (0.05 M), pH 12.5 NaOH solution (0.05 M), pH 3 phosphate buffer (0.05 M) titrated with HNO3. The concentration of the Ir atom in the stocked or diluted colloid solution was measured by the inductively coupled plasma mass spectrometry (ICP-MS) technique. The acidity/alkalinity of the solution was measured by a pH meter (AG, Sartorius, Göettingen, Germany). The morphology of the electrode surface was examined using a field emission scanning electron microscope (JSM-7000F, JEOL, Tokyo, Japan).3. Results and discussion
As can be seen in the cyclic voltammogram (curve a) of Fig. 1A, the PAH/IrOx·nH2O colloid assembled GCE exhibits anodic response conspicuously indicating the oxygen evolution at the potential > 0.8 V (versus Ag/AgCl) in a 0.05 M phosphate buffer (pH 7.2). The current reached 0.5 mA cm−2 at 0.87 V (versus Ag/AgCl). In comparison with a PAH-functionalized (curve b) and a bare GCE (curve c), the PAH/IrOx·nH2O colloid assembled GCE shows coupled redox peaks at 0.2 and 0.15 V, which are assigned to the IrIV/IrIII redox behavior (inset of Fig. 1A). PAH plays a decisive role in the production of a stable IrOx·nH2O film by means of a formation of the positively charged species, which is electrostatically pre-interacted with the negatively-charged carboxyl derivative established on the GCE. Note that neither the bare nor PAH-functionalized GCEs are capable of producing such a stable electrochemical response given with high OER catalytic merit, which thus demonstrates the applicability of PAH/IrOx·nH2O colloid assembled GCE.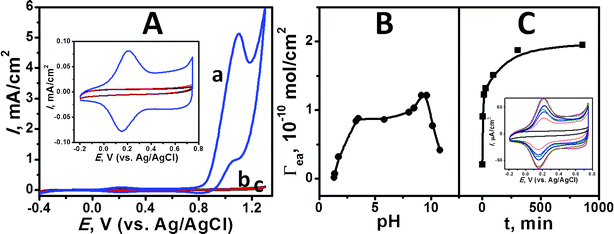 | ||
| Fig. 1 (A) Cyclic voltammograms (CVs) of (a) an iridium oxide colloid assembled (Γea = 1.89 × 10−10 mol cm−2), (b) PAH-functionalized, and (c) bare glassy carbon electrodes in a 0.05 M phosphate buffer (pH 7.2) measured in the potential ranging from −0.4 to 1.3 V (vs. Ag/AgCl) at 50 mV s−1. Inset illustrates the corresponding CVs in the potential range of −0.2–0.75 V. Effects of pH (B) and treated time (C) upon the electroactive amount (Γea) of the electroactive Ir sites adsorbed on the glassy carbon electrode in 0.18 mM Ir-containing solution. The treated time in (B) is 30 min. pH of the colloidal solution in (C) is 9.3. Inset in (C) shows CVs of iridium oxide colloid assembled electrode treated for 0, 5, 15, 30, 95, 305, and 860 mins in 0.18 mM Ir-containing solution at pH 9.3. | ||
The amount of electroactive Ir sites (Γea), based on the molar unit, was evaluated from the anodic peak area (excluding the blank capacitive current) at 0.2 V on the cyclic voltammogram. Fig. 1B and C depicts the dependence of Γea on the pH of the colloidal solution and treated time, respectively. After the PAH-functionalized GCE was treated with IrOx·nH2O colloidal solution in connection to varied pH through desired treatment period, cyclic voltammogram of the treated electrode was recorded in a 0.05 M phosphate buffer (pH 7.2). Over pH 1–11, Γea rose sharply from pH 1 to pH 3.5 and increased slightly at pH 4 to 8.5. The maximum amount reached 1.21 × 10−10 mol cm−2 at pH 9–9.6, followed by a steep decrease at escalating pH as a result of a neutralization of PAH. On the other hand, Γea is significantly dependent on the time treated with the colloidal solution in the first 30 min and is saturated at approximately 1.89 × 10−10 mol cm−2 after 300 min. An IrOx·nH2O colloid-functionalized electrode treated for more than 14 h was therefore utilized in the OER study to ensure a reproducible production and consistent quantity of electroactive Ir sites.
The coverage (Γcov) of the IrOx·nH2O colloid on GCE treated for 30 and 860 min was also analyzed by ICP-MS technique to evaluate the fraction (Γea/Γcov) of produced electrodes. Notably, Γcov is substantially lower (one or two orders of magnitude) than those reported in relevant works.18,21,22 The present electrode shows a high Γea/Γcov (approximately 12.5%), indicating a comparable effectiveness to 16%,21 where the citrate ion stabilized IrO2 colloid was self-assembled on an ITO electrode. Such low Γea and Γcov with high Γea/Γcov may be interpreted in terms of the intermittent formation of PAH/IrOx·nH2O cluster, as inferred in the SEM results (Fig. 2A). Varying from prior works,21,22 where a densely IrO2 covered surface was obtained, it was revealed that a well-dispersed PAH/iridium oxide colloid deposition yielding a nanostructured cluster with 50–100 nm in size (inset in Fig. 2A) accounted for the high Γea/Γcov.
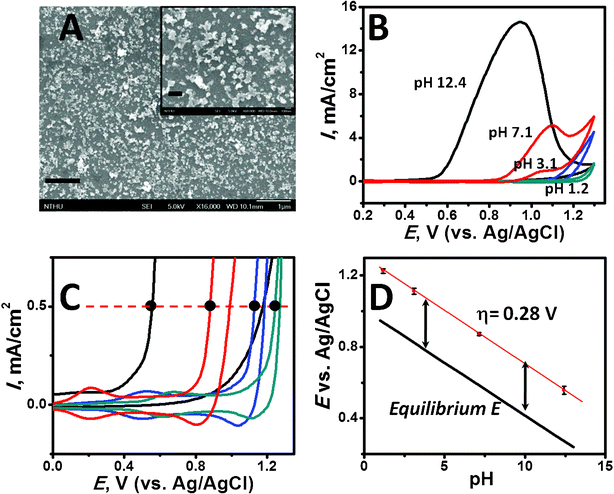 | ||
| Fig. 2 (A) SEM of an iridium oxide colloid adsorbed glassy carbon electrode assembled in 0.18 mM Ir-containing solution (pH 9.3) for 860 min. The scale bar is 1 μm. Inset: magnified image of (A) with a bar in 200 nm. (B) Cyclic voltammograms (50 mV s−1) of the iridium oxide colloid assembled (Γea = 1.89 × 10−10 mol cm−2) glassy carbon electrode in pH 1.2, 3.1, 7.1 and 12.1 solutions. (C) Magnified cyclic voltammograms at 0–1.3 V with black dots indicating the potential of initiation of water oxidation at 0.5 mA cm−2. (D) The overpotential (η) estimation given from a comparison of the acquired potentials (from C) to the pH-dependent equilibrium potential of the OER (black line). | ||
The OER catalytic capability of the produced iridium oxide colloid-assembled GCE electrode was also characterized. Fig. 2B illustrates the voltammograms at varied pHs. The sharply rising anodic currents at high potentials correspond to oxygen evolution. The waves reflect a mass transfer limited reaction at high potential as a result of low buffer capacity of the electrolyte.18,20The potential onset of the oxygen evolution at 0.5 mA cm−2 in the corresponding pH was recognized (shown as black dots in Fig. 2C) and the values are depicted in Fig. 2D, as dependence on pH. In comparison to the thermodynamic potentials of reaction 1 (depicted as black line in Fig. 2D), the results pointed out that the PAH/iridium oxide colloid assembled GCE electrode exhibited a nearly pH-independent η of 0.28 V for water oxidation. Furthermore, evaluating the site TOF of the PAH/iridium oxide colloid-assembled GCE by a ratio of currents at 0.87 V (four electrons/Ir at 0.28 V, to those at 0.2 V (one electron transfer of IrIV/IrIII) gave 6.36 ± 0.25 s−1 of site TOF in phosphate buffer (pH 7.2) (Fig. 3A), which is superior or comparable to the previously reported values.18,20–22 This result can be rationalized by the virtue of the effective catalytic sites given by the nanostructured cluster, albeit only ∼10−9 mol cm−2 Ir was deposited.
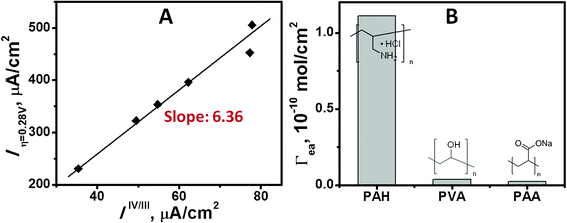 | ||
| Fig. 3 (A) Relationship between the currents at 0.87 (η = 0.28 V) and 0.2 V as a dependence on various Ir loading. The ratio of currents at 0.87 V (four electrons/Ir at η = 0.28 V) to those at 0.2 V (one electron transfer of to IrIV/IrIII) gives the Ir site TOF. (B) Effect of polymer under study on the electroactive Ir amount (Γea) adsorbed on the glassy carbon electrode. Concentration of polymer is 5 mg mL−1. The electrode is assembled in 0.18 mM Ir-containing solution (pH 9.3) for 15 min. Procedures to prepare the PVA- and PAA-functionalized electrodes are same with PAH, as illustrated in Experimental section. | ||
To elucidate and recognize the factor yielding the high OER catalytic capability in connection to a low Ir loading, a series of experiments, described below, were undertaken for gaining better understanding of catalytic characterization and to identify the active species. Despite iridium oxide films that are recently made by anodic deposition and by particle assembly in which either IrOx·nH2O or IrO2 is deemed as the functional component catalytic to OER, the ligand-free IrOx·nH2O nanoparticle is considered the neutral species that is least preferred to electrostatically interact with the positively charged PAH. We therefore hypothesize that the hexahydroxyiridate [Ir(OH)6]2−, one of the main constituents in the colloidal solution, plays the catalytic role in OER.
In order to investigate the functionality of positively charged amine group in PAH, polyvinyl alcohol (PVA) and polyacrylic acid (PAA), both possessing vinyl backbones in the monomeric unit with hydroxyl- (neutral) and carboxyl- (negatively charged) derivatives, were employed in the replacement of PAH. The electrode was prepared as per the regular protocols, with 5 mg mL−1 PVA or PAA in 0.05 M NaCl solution. Fig. 3B depicts the Γea as dependence on the polymer under investigation. It was revealed that the electrode assembled with both PVA and PAA adsorbed little active Ir (3.85 × 10−12 and 2.49 × 10−12 mol cm−2, respectively) inferring that the positively charged amine group indeed played crucial role in adsorbing iridium oxide colloid. Moreover, obviating the possibility of forming a covalent bond between the amine (on PAH) and Ir–O–Ir linkage (on IrOx) accounts for the likely electrostatic attraction between [Ir(OH)6]2− and PAH.
Mallouk and coworkers20 illustrated an IrOx-free precursor, prepared at pH 8–9. Such a precursor, composed of highly concentrated [Ir(OH)6]2−, was used in this study to elucidate the active species assembled on the PAH-functionalized GCE. The cyclic voltammogram obtained from the PAH-functionalized GCE was treated with the precursor (at pH 8.2), illustrated in Fig. S2†, indicating that a comparable signal (2.73 μA of the anodic peak current converting Ir(III) to Ir(IV)) to the result observed at similar pH (3.11 μA measured at pH 8 shown in Fig. 1B). This result manifests a feasible formation of the iridium oxide colloid assembled electrode from the [Ir(OH)6]2−-containing solution and its catalytic capability towards OER.
We thus envisaged the electrostatic interaction between the iridium oxide colloid and PAH at pH 1, 8–9, and 12, as shown in Fig. 4. In an acidic condition, the colloidal solution IrOx·nH2O nanoparticles are plentiful, ascribable to the acidic condensation19, and the PAH is highly protonated, yielding high fractions of the positively charged amine group. Little [Ir(OH)6]2− is present in the colloidal suspension, resulting in a low Γea (as shown in Fig. 1B). On the other hand, substantial IrOx·nH2O is hydrolyzed to [Ir(OH)6]2− in the basic condition (e.g., pH 12), while the PAH is deprotonated to create a detrimental circumstance towards the assembly of [Ir(OH)6]2−. Maximum assembly of [Ir(OH)6]2− onto the PAH-functionalized electrode was observed at pH 8–9 as a result of fractional composition of [Ir(OH)6]2− and IrOx·nH2O, as well as the partial protonation of amine moiety of PAH to expedite the electrostatic assembly between [Ir(OH)6]2− and PAH. Although a majority of prior works18–20,23 considered IrOx·nH2O nanoparticles as the catalytic component towards OER, significant endeavors have been made to demonstrate that the deposited iridium complex [Cp*Ir(H2O)3]2+ (from the molecular precursor) undertakes a heterogeneous water oxidation with oxygen produced as the product.24
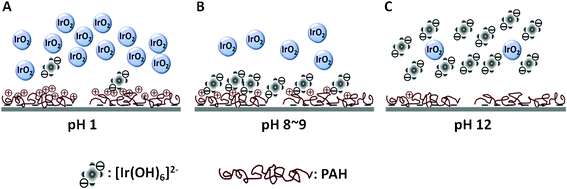 | ||
| Fig. 4 Envisaged illustration of the electrostatic interaction of iridium oxide colloid upon the glassy carbon electrode at pH 1 (A), 8–9 (B), and 12 (C). | ||
Conclusions
In conclusion, we utilized a positively charged polymer to facilitate the formation of an actively electrocatalytic PAH/iridium oxide colloidal deposition on the glassy carbon electrode via an electrostatic assembly fashion. As such, the produced electrode yields a high Γea/Γcov of 12.5% in connection to a minimal coverage (2.3 × 10−9 mol cm−2 of Γcov) reflected from the well-dispersed nanostructured deposition of PAH/iridium oxide colloid cluster, leading to a high site turnover efficiency of 6.36 ± 0.25 s−1. Our postulation was also verified in the current study that hexahydroxyiridate(IV) [Ir(OH)6]2− is the active catalyst towards OER. Pursuant to the minimal Ir loading yielding the similar turnover efficiency, the electrostatically assembled iridium oxide colloid deposition holds spectacular promise as an efficient catalyst of OER.Acknowledgements
This work is supported by National Science Council Taiwan under contracts 98-2113-M-002-025-MY3, 100-2113-M-002-016-MY2 and 100-2811-M-002-023. The authors would like to thank Professor Chun-Hsien Chen in Department of Chemistry, National Taiwan University for valuable discussions and suggestions.References
- Q. Schiermeier, J. Tollefson, T. Scully, A. Witze and O. Morton, Nature, 2008, 454, 816–823 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS.
- R. Brimblecombe, G. C. Dismukes, G. F. Swiegers and L. Spiccia, Dalton Trans., 2009,(43), 9374–9384 RSC.
- M. Yagi, A. Syouji, S. Yamada, M. Komi, H. Yamazaki and S. Tajima, Photochem. Photobiol. Sci., 2009, 8, 139 CAS.
- H. Yamazaki, A. Shouji, M. Kajita and M. Yagi, Coord. Chem. Rev., 2010, 254, 2483–2491 CrossRef CAS.
- G. Beni, L. Schiavone, J. Shay, W. Dautremont-Smith and B. Schneider, Nature, 1979, 282, 281–283 CrossRef CAS.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795–2806 RSC.
- K. Kalyanasundaram and M. Gratzel, Angew. Chem., Int. Ed. Engl., 1979, 18, 701–702 CrossRef.
- J. Kiwi and M. Gratzel, Angew. Chem., Int. Ed. Engl., 1978, 17, 860–861 CrossRef.
- A. Mills and N. McMurray, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 379–390 RSC.
- R. S. Chen, Y. S. Chen, Y. S. Huang, Y. L. Chen, Y. Chi, C. S. Liu, K. K. Tiong and A. J. Carty, Chem. Vap. Deposition, 2003, 9, 301–305 CrossRef CAS.
- M. El Khakani and M. Chaker, Thin Solid Films, 1998, 335, 6–12 CrossRef CAS.
- M. El Khakani, M. Chaker and E. Gat, Appl. Phys. Lett., 1996, 69, 2027 CrossRef.
- D. J. Comstock, S. T. Christensen, J. W. Elam, M. J. Pellin and M. C. Hersam, Electrochem. Commun., 2010, 12, 1543–1546 CrossRef CAS.
- J. Hu, M. Abdelsalam, P. Bartlett, R. Cole, Y. Sugawara, J. Baumberg, S. Mahajan and G. Denuault, J. Mater. Chem., 2009, 19, 3855 RSC.
- M. Yagi, E. Tomita and T. Kuwabara, J. Electroanal. Chem., 2005, 579, 83–88 CrossRef CAS.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958–12961 CAS.
- Y. Zhao, E. A. Hernandez-Pagan, N. M. Vargas-Barbosa, J. L. Dysart and T. E. Mallouk, J. Phys. Chem. Lett., 2011, 2, 402–406 CrossRef CAS.
- Y. Zhao, N. M. Vargas-Barbosa, E. A. Hernandez-Pagan and T. E. Mallouk, Small, 2011, 7, 2087–2093 CrossRef CAS.
- T. Kuwabara, E. Tomita, S. Sakita, D. Hasegawa, K. Sone and M. Yagi, J. Phys. Chem. C, 2008, 112, 3774–3779 CAS.
- M. Yagi, E. Tomita, S. Sakita, T. Kuwabara and K. Nagai, J. Phys. Chem. B, 2005, 109, 21489–21491 CrossRef CAS.
- T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578–15579 CrossRef CAS.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473–10481 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra00895e/ |
| This journal is © The Royal Society of Chemistry 2012 |