One-pot synthesis of a polyaniline–silver nanocomposite prepared in ionic liquid
Cintia Marques
Correa
,
Roselena
Faez
,
Marcos Augusto
Bizeto
and
Fernanda Ferraz
Camilo
*
Instituto de Ciências Ambientais, Químicas e Farmacêuticas, Universidade Federal de São Paulo, Rua Artur Riedel, 275 – CEP: 09972-270 – Diadema, SP, Brazil. E-mail: ffcamilo@unifesp.br; Fax: +55 11 4059-3618; Tel: +55 11 4059-3618
First published on 21st February 2012
Abstract
A mixture of aniline and a silver salt in ionic liquid has afforded a composite of PANI, in its more conducting form, and silver nanoparticles through a single-step direct reaction, without addition of any acid species or template. An ionic liquid acted as the solvent and template for the nanostructured material. The silver salt (AgTf2N) used in this work is soluble in the chosen ionic liquid, which is not an usual characteristic. TEM analysis of the obtained composite showed the presence of rounded silver nanoparticles with average diameter size around 10–20 nm uniformly dispersed in the polymeric matrix. XRD corroborates the size of the metallic particles at the nanoscale. FTIR and UV-vis confirmed that PANI was obtained in emeraldine salt form. The composite is slightly soluble in DMSO, an unusual feature for bulk conducting polyaniline. The composite shows electrical conductivity 100 times higher than bulk PANI prepared by usual procedures and its electroactivity was studied by cyclic voltammetry. Therefore, in this study it was shown that Ag+, in ionic liquid, is able to quickly oxidize the aniline to PANI, which is not observed in another media.
Introduction
Polyaniline (PANI) is one of the most studied conducting polymers due to its easy preparation method, chemical and thermal stabilities and relatively high electrical conductivity. PANI has demonstrated many potential applications in electronic or optical devices and chemical or biological sensors.1–5 The embedding of metallic material in PANI, mainly at the nanoscale, is one attractive strategy to enhance its sensing and catalytic capabilities and to change its electrical and optical features.6–11Succinctly, there are two general methods to produce nanocomposites of conducting polymers and metals. In the first one, metal nanoparticles are added directly to the polymeric matrix, but with the major inconvenience of promoting the agglomeration of the inorganic phase.12–13 In the second, nanoparticles are generated during the polymerization step in order to afford a more homogeneous material with uniform distribution of the nanoparticles.14–16
Among the metal–PANI composites, those with silver have been extensively investigated and their preparation from the direct oxidation of aniline with silver cations seems to be the simplest manner. However, taking into account that polyaniline is prepared by oxidation with ammonium persulfate in acid medium (the standard reduction potential of S2O82− + 2e− → 2 SO42− is 2.01 V vs. NHE), it is hard to consider that Ag+ is the oxidant agent in the early stages of the polymerization of the monomer, due to its lower standard reduction potential (Ag+ + e− → Ag is 0.79 V vs. NHE) than the aniline one (around 1.0 V vs. NHE).17 To circumvent such aspects, several groups are trying different strategies to use the silver cation to oxide aniline, such as heating,18 irradiation with UV-vis,19γ radiation17 and ultrasound in aqueous media.20 In the absence of these conditions, the reaction occurs slowly, taking weeks and even months to complete.17,21 Although preparations of composites of PANI and silver have already been reported,17–27 the development of methods to produce them quickly and with high yield is still a challenge.
Ionic liquids (ILs) are alternative solvents that have been used in a wide range of synthetic applications due to their unique physical properties including non-flammability and negligible vapor pressure.28 Their intrinsic ionic conductivity and good electrochemical stability categorize them as alternative electrolytes for electrochemical devices29 and as medium to prepare conducting polymers.30,31 In addition, their ionic charge, high polarity and dielectric constant and supramolecular network provide a unique medium for the preparation of nanomaterials without the need of any extra stabilizing molecules.32–38
In 2008, Pringle and co-workers39 reported the synthesis of conducting polymers (poly(terthiophene) and polypyrrole) with gold and silver nanoparticles, in ionic liquids using gold chloride and silver nitrate as oxidant agents.39 Due to the low solubility of the silver salt in the ionic liquid, a high volume of ionic liquid and heating were required to afford a homogeneous medium.
Herein, we describe a one-pot synthesis of polyaniline–silver (PANI/Ag) composite starting from aniline and a silver salt ionic liquid solution (AgTf2N in BMITf2N, see Scheme 1). No acid was added in this preparation and an ionic liquid was used as solvent and template at the same time for the nanocomposite formation. The motivation of this work was to propose an one step chemical reaction between two non conductive species in a green solvent to produce a nanocomposite with high conductivity and uniformity. Since the polymer and silver nanoparticles are produced simultaneously in a homogeneous phase, it is expected to observe an intimate contact between the organic and inorganic phases.
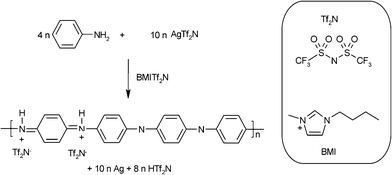 | ||
| Scheme 1 Preparation of PANI/Ag nanocomposite and the ionic liquid chemical structure used in this work. | ||
Experimental section
Materials
Aniline (ALDRICH, 97%) was distilled twice under reduced pressure before use. Other chemicals were used without previous purification. BMITf2N was prepared following a procedure already described in the literature.40Preparation
Preparation of AgN(SO2CF3)2 (AgTf2N)
Into a 50 mL vacuum sublimator were added 5.14 g (18.0 mmol) of anhydrous LiN(SO2CF3)2 (ALDRICH, 97%) followed by 25 mL of H2SO4 (ALDRICH, 98%). The solution was heated at 70 °C whilst stirring, and HN(SO2CF3)2 produced was collected in the cooler part of the sublimator as a white solid (90% yield). In the next step, a slight excess of Ag2CO3 (12.0 mmol, newly synthesized) was added, under stirring, to a solution of HN(SO2CF3)2 (5.62 g; 20.0 mmol) in 50 mL of distilled water. The reaction mixture was heated at 65 °C and then a white solid was isolated by removing water under reduced pressure. To remove impurities, such as silver oxide, the solid was dissolved in 50 mL of dry diethyl ether and the solution was filtered. After solvent removal under reduced pressure, AgN(SO2CF3)2 was obtained as white powder with 70% yield.Preparation of PANI/Ag composite
451 mg (4.86 mmol) of aniline was added, under stirring, to 4.70 g (12.1 mmol) of AgN(SO2CF3)2 dissolved in 6.0 mL of BMITf2N. In a few minutes, the reaction mixture turns green. After 24 h, the precipitate was isolated by filtration, rinsed with acetone and dried under reduced pressure. PANI/Ag (1.21 mg) was obtained as a green solid (yield = 80%, considering 30% of organic content determined by thermogravimetry analysis and the initial mass of the monomer).Characterization
Thermogravimetric analysis (TG) was performed in a TGA DUPONT thermogravimetric analyzer, using air atmosphere (100 cm3 min−1) and alumina capsule at a heating rate of 20 °C min−1 in the temperature range of 20 °C to 900 °C. Differential scanning calorimetry analysis (DSC) was performed in DSC DUPONT equipment, using dry nitrogen atmosphere (300 cm3 min−1) and platinum crucible at a cooling rate of 20 °C to −130 °C, maintained at −130 °C for 10 min, then at a heating rate of 10 °C min−1 in the range 20 °C to 300 °C.Mass coupled thermogravimetric analysis (TG-MS) was performed on a NETZSCH THERMOANALYSER model TGA/DSC 490 PC Luxx coupled to an Aëolos 403C mass spectrometer, using a heating rate of 10 °C min−1 and synthetic air flow of 50 mL min−1.
UV-visible electronic absorption spectra (UV-vis) of solid samples were recorded in a Shimadzu spectrophotometer model UV-2401PC equipped with an integration sphere using 25 mg of the samples diluted in 500 mg of BaSO4 which is also the reference. The spectra were recorded in reflectance mode and transformed to absorbance using the equipment software (Kubelka-Munk method option).
The electrical conductivity was measured by a four-point method using a Jandel Multi Height Probe with Jandel cylindrical probe head (25.4 nm diameter × 48.5 nm high), tip spacing of 1.591 nm controlled by the RM3-AR Test Unit. The pressed pellets (15 mm in diameter and 0.50 mm in thickness, respectively) used in this measurement were prepared using 100 mg of the sample. Electrical conductivity of the pressed pellet was an average of eight values measured from two sides and in different places and it was determined for two different pellets.
X-ray diffraction (XRD) patterns of powdered samples were recorded on a Rigaku diffractometer model Miniflex using Cu-Kα radiation (1.541 Å, 30 kV and 15 mA).
1H NMR spectra were recorded on a Bruker AC-300 spectrometer using DMSO-d6 as the solvent and internal standard.
FTIR spectra were recorded on a Perkin-Elmer 1750 series grating in the range of 400–4000 cm−1. Samples were dispersed in potassium bromide and compressed into pellets.
TEM micrographs were recorded on a JEM 1200 EX II (JEOL) operating at 80 kV. Samples were prepared by dispersing the solids in isopropanol with an ultrasonic bath followed by deposition on a carbon-coated Cu microgrid.
Cyclic voltammetric measurements were performed with an Autolab/PGSTAT-30 (Eco Chemie) potentiostat/galvanostat connected to a microcomputer. ITO substrates with a resistivity of 20 Ω (Delta Technologies Company) were used and cleaned by using acetone in an ultrasonic bath for 10 min before use. The experiments were carried out in a single compartment electrochemical cell, using Pt as counter electrode and a Ag/AgCl (saturated with KCl) reference electrode. The polymeric film was deposited on the electrode (ITO) by drop casting, using a PANI/Ag solution in DMSO (20 mg of the composite in 2 mL of DMSO) and Ag/AgCl as the reference electrode.
Results and discussions
The PANI/Ag composite was prepared using a one step reaction procedure using aniline as the monomer and a 2 M solution of AgTf2N in BMITf2N, according to Scheme 1. The reaction mixture colour changed from colourless to yellow, then brown and finally deep green in 1 h. After 24 h, the crude product was isolated as a green-silver powder with 80% yield. The material is slightly soluble in DMSO, producing a green solution and it is not soluble in acetone, ethanol, dichloromethane or chloroform.The obtained nanocomposite was investigated by FTIR spectroscopy (Fig. 1), and bands were observed in the spectrum at 1588 and 1492 cm−1, which were assigned to quinoid and benzenoid stretching modes. Bands around 2920 and 2850 cm−1 were attributed to C–H stretching vibrations and the band at 1139 cm−1 was assigned to in plane C–H bending vibrations in the quinoid rings of the doped PANI. The broad band in the region around 3400 cm−1 corresponded to N–H bond stretching. All these bands are coherent with the emeraldine salt form of PANI.41 The asymmetric stretching CF3 and S–N–S bands near 1200 cm−1 and 1056 cm−1, respectively, and the symmetric stretching SO2 band close to 1339 cm−1 confirm that Tf2N is the counterion of PANI.42
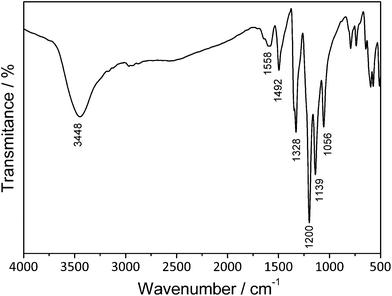 | ||
| Fig. 1 FTIR spectrum of the PANI/Ag composite in a KBr pellet. | ||
Comparing the UV-vis spectra of the PANI/Ag composite and pure PANI (Fig. 2) recorded in the solid state, one band around 800 nm with a free carrier tail extending into the near-infrared region can be observed as a common feature, which is attributed to π-polaron transition and is characteristic of emeraldine salt.43 The higher intensity of the band centred at 405 nm and the small shift to a lower wavelength compared to the UV-vis spectrum of pure PANI (Fig. 2) can be explained by the overlapping of the absorption band assigned to the polaron-π* transition (usually centred at 440 nm for pure PANI44) and the surface plasmon resonance absorption of the electrons in the conducting silver bands at 410 nm.
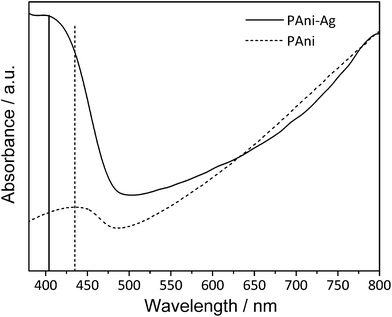 | ||
| Fig. 2 UV-vis spectra of the PANI/Ag composite and pure PANI. | ||
The thermal stability of the nanocomposites was evaluated by thermogravimetric analysis (TGA) (Fig. 3). The first weight loss event in the range of 25–390 °C was attributed to the initial removal of the dopant, Tf2N anion, and the second due to the thermal decomposition of the polymer backbone chains. Confirmation of the Tf2N removal as the first step was performed by coupling TGA to a mass spectrometer, which allows the identification of the evolved decomposition gases. The inset in Fig. 3 shows the mass curves of the SO (m/z = 48) and SO2 (m/z = 64) fragments in the range 200–400 °C, which is coherent with species produced in the decomposition of the CF3–SO2–N–SO2–CF3 structure. TGA was also a suitable method for the determination of silver content in the nanocomposite, which corresponds to around 70% of the composite mass.
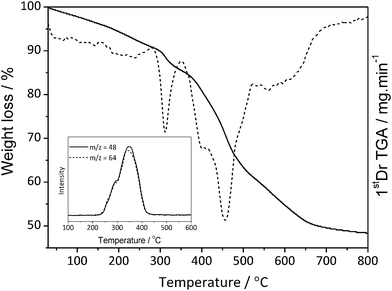 | ||
| Fig. 3 TGA curves of the PANI/Ag composite obtained under air atmosphere at a heating rate of 20 °C min−1. | ||
Fig. 4 shows the DSC curve of the PANI/Ag composite. The sharp endothermic peak at around 190 °C is not observed in bulk PANI45 and, according to the TGA curve, there is no weight loss associated with it. This process corresponds to the melting process of the silver nanoparticles.46
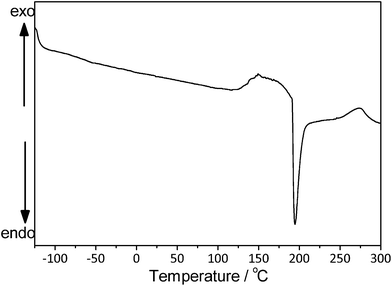 | ||
| Fig. 4 DSC curve of the PANI/Ag composite obtained under dry nitrogen atmosphere and a heating rate of 10 °C min−1. | ||
PANI is considered insoluble in several solvents, which hampers its characterization and processability in solution state. Due to the slight solubility of the PANI/Ag composite prepared in this work, the 1H NMR spectrum of this composite was registered in DMSO-d6, as shown in Fig. 5. The spectrum has a broad peak in the range delta of 6.50–8.00 ppm due to the combined multiplets of protons from benzenoid and quinoid rings of the emeraldine salt.
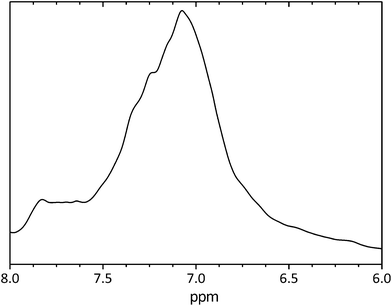 | ||
| Fig. 5 1H NMR spectrum of the PANI/Ag composite in DMSO-d6. | ||
The registered XRD pattern of the PANI/Ag composite (Fig. 6) presents two intense diffraction peaks at 38° and 44° that are attributed to (111) and (200) planes of the face-centered cubic phase of silver, which were very close to that given by the ICDD file n° 4–0783. Taking into account the most intense diffraction peak, the average silver particle size was determined using the Scherrer equation, considering the shape factor (K) as spherical (0.9). This calculation resulted in particles with estimated average size around 17 nm. The structure of PANI varies from amorphous to partially crystalline depending on the polymer doping state.47 The most crystalline phase occurs when the PANI is in the emeraldine salt form. The polymer in this form presents diffraction peaks at 20.4 and 24.8°, which are respectively ascribed to the parallel and perpendicular periodicity of PANI chains.47 The inset of Fig. 6 shows a magnification of the PANI diffraction peak region where a diffraction halo is observed with a maximum at around 21°, however, due to the high intensity of the Ag diffraction peaks it is not possible to infer properly the crystallinity of the PANI.
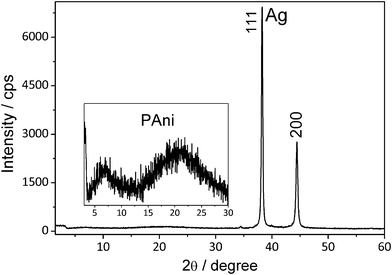 | ||
| Fig. 6 Powder XRD pattern of the PANI/Ag composite. Inset: enlargement of the region of the PANI diffraction peaks. | ||
The PANI/Ag nanocomposite was examined by TEM as shown in Fig. 7. The registered micrographs indicate that the Ag particles produced are in the nanoscopic domain as previously indicated by the X-ray diffractometry. The particle size histogram registered from Fig. 7C indicates that the average diameter is in the range of 10–20 nm. It is possible to observe in the micrographs that the nanoparticles have a rounded shape and are well dispersed in the polymeric matrix (see Fig. 7A). This kind of arrangement will impact the electronic conductivity of the material since the metal particles will not be in direct contact.
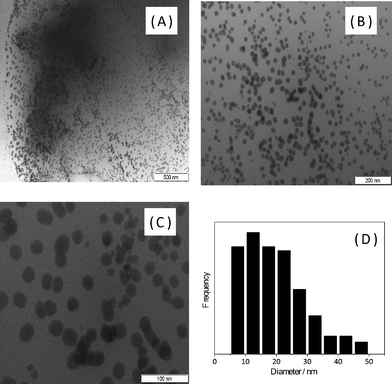 | ||
| Fig. 7 TEM micrographs of the PANI/Ag composite registered in different magnifications (A, B, C) and particle size histogram (D). | ||
Since Ag is a good electrical conductor, the conductivity of the nanocomposite should be improved by dispersion of the noble metal in PANI, thus providing a more effective electrical pathway. At room temperature, electrical conductivity of the composite was evaluated in the order of 100 S cm−1. Comparing these value with that obtained for bulk PANI (1–5 S cm−1),48 there is an increase of about two orders of magnitude in the conductivity upon incorporation of Ag nanoparticles in the conductive form of polyaniline using the present method. This increase is rather poor, considering the high content of silver in the composite, however this is normal behaviour for composites of conducting polymers and metals, where the distribution of the metallic material in the polymeric matrix is homogeneous and the conductivity is governed by the organic polymer. It is worth to note that the conductivity values reported so far for PANI/Ag in the literature have never exceeded 101 S cm−1, except in two cases where the conductivity reached the order of 103 S cm−1.21,24
Cyclic voltammetry is one of the most useful techniques to evaluate the electro-activity of polyaniline. A cyclic voltammogram of a film of the PANI/Ag nanocomposite, deposited by casting on ITO performed in 0.1 M HCl solution at different scan rates is shown in Fig. 8A.
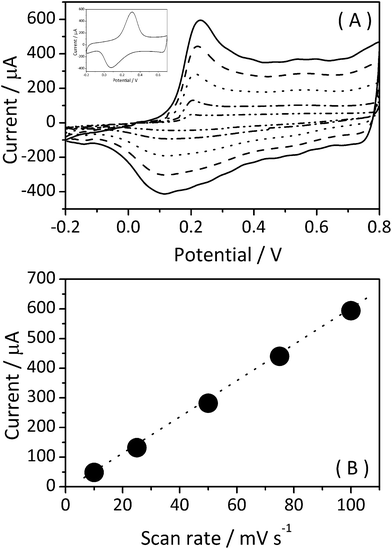 | ||
| Fig. 8 (A) Cyclic voltammograms of the PANI/Ag composite in aqueous HCl 0.1 M solution at different scan rates (from inner to outer curve: 10, 25, 50, 75 and 100 mV s−1). Inset: cyclic voltammogram of pure PANI in aqueous HCl 0.1 M solution at 50 mV s−1. (B) Intensity of the cathodic peak as a function of scan rate. | ||
The voltammograms show clearly one reversible redox process with a cathodic peak at around 0.25 V and one anodic peak at 0.1 V, corresponding to the characteristic transformation of the leucoemeraldine form to the emeraldine form of pure PANI (inset in Fig. 8A). During the voltammetric analysis, the composite changed its color, typically like the electrochromism observed in PANI. It is interesting to note that there are no signals due to the oxidation of the Ag in the studied conditions, suggesting that these nanoparticles are surrounded by polymeric material. Also, the poor definition between the two peaks in any scan rate observed in the CVs suggests strong interaction between the polymer and metallic nanoparticles. When the scan rate was varied to 10, 25, 50, 75 and 100 mV s−1, the cathodic and anodic peak potentials increase linearly with the scan rate, indicating that the electrode process is a surface-controlled process (Fig. 8B).
Conclusions
In summary, in this paper a one-pot synthetic procedure to obtain a nanocomposite of Ag/PANI was shown, which contains the polymer in its more conductive form and silver nanoparticles with an average diameter of less than 20 nm. The procedure was based on a simple oxidation reaction of aniline by silver cations, which act as the oxidant and the metal source of the nanoparticles at the same time. This reaction was able to occur due to the use of an ionic liquid both as a solvent and the template for the nanocomposite formation. The resulting composite presents enhanced electrical conductivity, an increase of about two orders of magnitude compared to bulk PANI. In ionic liquid, Ag+ was able to quickly oxidize aniline to PANI, since its oxidant power is dependent on the medium (solvent) and on its counterion.49 Finally, it is worth mentioning that the product does not need to be isolated from the reaction mixture, since it is constituted of only a polymer, metallic particles and a secure solvent.Acknowledgements
The authors thank FAPESP (2007/50742-2) for the financial support. We also acknowledge CESQ-POLI-USP for the TGA and DSC analyses, Prof. Vera R. L. Constantino (IQ-USP) for the X-ray diffractometry, UV-vis and TG-MS analyses and Centro de Microscopia Eletrônica do Instituto BUTANTAN for the TEM analysis.References
- A. J. Heeger, J. Phys. Chem. B, 2001, 105, 8475 CrossRef CAS.
- J. Jang, J. Ha and J. Cho, Adv. Mater., 2007, 19, 1772 CrossRef CAS.
- Z. Zhang, J. Sui, L. Zhang, M. Wan, Y. Wei and L. Yu, Adv. Mater., 2005, 17, 285 CrossRef.
- J. Han, G. Song and R. Guo, Adv. Mater., 2007, 19, 299 Search PubMed.
- E. M. Genies, A. Boyle, M. Lapkowski and C. Tsintavis, Synth. Met., 1990, 36, 139 CrossRef CAS.
- R. A. Barros, C. R. Martins and W. M. Azevedo, Synth. Met., 2005, 155, 35 CrossRef.
- D. Y. Shin and I. Kim, Nanotechnology, 2009, 20, 415301 CrossRef.
- A. Kitani, T. Akashi, K. Sugimoto and S. Ito, Synth. Met., 2001, 121, 130 CrossRef.
- A. Drelinkiewicz, M. Hasik and M. Kloc, Catal. Lett., 2000, 64, 41 CrossRef CAS.
- P. T. Radford and S. E. Creager, Anal. Chim. Acta, 2001, 449, 199 CrossRef CAS.
- Y. Gao, D. Shan, F. Cao, J. Gong, X. Li, H. Y. Ma, Z. M. Su and L. Y. Qu, J. Phys. Chem. C, 2009, 113, 15175 CAS.
- A. Choudhury, Sens. Actuators, B, 2009, B138, 318 CrossRef CAS.
- Y. Leroux, E. Eang, C. Fave, G. Trippe and J. C. Lacroix, Electrochem. Commun., 2007, 9, 1258 CrossRef CAS.
- M. Lee, B. W. Kim, J. D. Nam, Y. Lee, Y. Son and S. J. Seo, Mol. Cryst. Liq. Cryst., 2003, 407, 397 CAS.
- P. K. Khanna, N. Singh, S. Charan and A. K. Viswanath, Mater. Chem. Phys., 2005, 92, 214 CrossRef CAS.
- M. R. Karim, K. T. Lim, C. J. Lee, M. T. I. Bhuiyan, H. J. Kim, L. S. Park and M. S. Lee, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 5741 CrossRef CAS.
- S. K. Pillalamarri, F. D. Blum, A. T. Tokuhiro and M. F. Bertino, Mater. Lett., 2005, 17, 5941 CAS.
- J. Du, Z. Liu, B. Han, Z. Li, J. Zhang and Y. Huang, Microporous Mesoporous Mater., 2005, 84, 254 CrossRef CAS.
- J. Li, H. Tang, A. Zhang, X. Shen and L. Zhu, Macromol. Rapid Commun., 2007, 28, 740 CrossRef CAS.
- R. A. Barros and W. M. Azevedo, Synth. Met., 2008, 158, 922 CrossRef.
- N. V. Blinova, J. Stejskal, M. Trchova, I. Sapurina and G. Ciric-Marjanovic, Polymer, 2009, 50, 50 CrossRef CAS.
- K. Gupta, P. C. Jana and A. K. Meikap, Synth. Met., 2010, 160, 1566 CrossRef CAS.
- W. M. Azevedo, R. A. Barros and E. F. Silva Jr, J. Mater. Sci., 2008, 43, 1400 CrossRef CAS.
- N. V. Blinova, P. Bober, J. Hromadkova, M. Trchova, J. Stejskal and J. Prokes, Polym. Int., 2010, 59, 437 CrossRef CAS.
- S. Bouazza, V. Alonzo and D. Hauchard, Synth. Met., 2009, 159, 1612 CrossRef CAS.
- A. B. Afzal, M. J. Akhtar, M. Nadeem, M. Ahmad, M. M. Hassan, T. Yasin and M. Mehmood, J. Phys. D: Appl. Phys., 2009, 42, 01541 CrossRef.
- J. Stejskal, M. Trchova, J. Kovarova, L. Brozova and J. Prokes, React. Funct. Polym., 2009, 69, 86 CrossRef CAS.
- T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- A. Fernicola, B. Scrosati and H. Ohno, Ionics, 2006, 12, 95 CrossRef CAS.
- W. Lu, A. G. Fadeev, B. Qi, E. Smela, B. R. Mattes, J. Ding, G. M. Spinks, J. Mazurkiewicz, D. Zhou, G. G. Wallace, D. R. MacFarlane, S. A. Forsyth and M. Forsyth, Science, 2002, 297, 983 CrossRef CAS.
- F. F. C. Bazito, L. T. Silveira, R. M. Torresi and S. I. C. de Torresi, Phys. Chem. Chem. Phys., 2008, 10, 1457 RSC.
- J. Dupont, G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner and S. R. Teixeira, J. Am. Chem. Soc., 2002, 124, 4228 CrossRef CAS.
- G. S. Fonseca, G. Machado, S. R. Teixeira, G. H. Fecher, J. Morais, M. C. M. Alves and J. Dupont, J. Colloid Interface Sci., 2006, 301, 193 CrossRef CAS.
- C. W. Scheeren, G. Machado, S. R. Teixeira, J. Morais, J. B. Domingos and J. Dupont, J. Phys. Chem. B, 2006, 110, 13011 CrossRef CAS.
- K. S. Kim, S. Choi, J. H. Cha, S. H. Yeon and H. Lee, J. Mater. Chem., 2006, 16, 1315 RSC.
- J. Zhu, Y. Shen, A. Xie, L. Qiu, Q. Zhang and S. Zhang, J. Phys. Chem. C, 2007, 111, 7629 CAS.
- K.-I. Okazaki, T. Kiyama, K. Hirahara, N. Tanaka, S. Kuwabata and T. Torimoto, Chem. Commun., 2008, 691 RSC.
- M. A. Gelesky, A. P. Umpierre, G. Machado, R. R. B. Correia, W. C. Magno, J. Morais, G. Ebeling and J. Dupont, J. Am. Chem. Soc., 2005, 127, 4588 CrossRef CAS.
- J. M. Pringle, O. Winther-Jensen, C. Lynam, G. G. Wallace, M. Forsyth and D. R. MacFarlane, Adv. Funct. Mater., 2008, 18, 2031 CrossRef CAS.
- K. Bonhote, A. P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Graetzel, Inorg. Chem., 1996, 65, 1168 CrossRef.
- S. Quillard, K. Berrada, G. Louarn, S. Lefrant, M. Lapkowski and A. Pron, New J. Chem., 1995, 19, 365 CAS.
- M. J. Muldoon and C. M. Gordon, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3865 CrossRef CAS.
- J. Stejskal, P. Kratochvil and N. Radhakrishnan, Synth. Met., 1993, 61, 225 CrossRef CAS.
- A. B. Afzal and M. Javed Akhtar, J. Inorg. Organomet. Polym. Mater., 2010, 20, 783 CrossRef CAS.
- K. C. Yung, S. P. Wu and H. Liem, J. Mater. Sci., 2009, 44, 154 CrossRef CAS.
- N. H. Kim, J.-H. Kim and K. K. Ihn, J. Nanosci. Nanotechnol., 2007, 7, 3805 CrossRef CAS.
- L. Zhang and M. Wan, Adv. Funct. Mater., 2003, 13, 815 CrossRef CAS.
- J. Stejskal, Pure Appl. Chem., 2002, 74, 857 CrossRef CAS.
- G. N. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |