ESI-MS mechanistic studies of Wacker oxidation of alkenes: dinuclear species as catalytic active intermediates†
Dominique
Harakat
,
Jacques
Muzart
and
Jean
Le Bras
*
Institut de Chimie Moléculaire de Reims-UMR 7312 CNRS-Université de Reims Champagne-Ardenne, UFR des Sciences exactes et naturelles, BP 1039, 51687 REIMS Cedex 2, France. E-mail: jean.lebras@univ-reims.fr
First published on 23rd February 2012
Abstract
The monitoring, by ESI-MS, of the Wacker oxidation of alkenes, using benzoquinone as terminal oxidant, has shown that dinuclear palladium complexes are more involved as active catalytic intermediates than mononuclear species. Kinetic experiments have confirmed a palladium-dependency greater than first-order, pointing out that the catalytic active species are formed by an associative process. The ESI-MS analysis associated with tests of complexation has also shown that the reoxidation of Pd occurs before the decoordination of the product.
Introduction
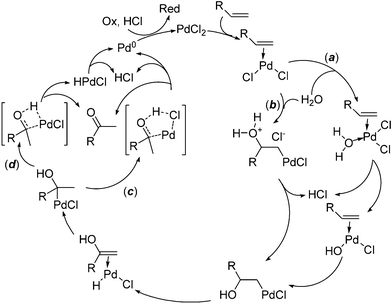
The Pd(II)-catalyzed oxidation of alkenes with water, the so-called Wacker reaction, has been the subject of intense studies and has extensive synthetic applications.1 The classical conditions in organic synthesis, also referred to as Tsuji–Wacker conditions, use N,N-dimethylformamide (DMF)/H2O mixtures and oxidants such as copper salts/O2 or p-benzoquinone (BQ).2 The mechanism of such transformations has been investigated through isotope effects, kinetic, stereochemical, and theoretical studies.3 The simplified mechanism shown in Scheme 1 involves alkene coordination to the palladium(II) (e.g. PdCl2), followed by hydroxypalladation; its stereochemistry, syn (path a) or anti (path b), depends on the experimental conditions. β-Hydride elimination affords a palladium–enol π-complex, which evolves to a Pd–alkyl complex via insertion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond into H–Pd. A halide-mediated reductive elimination (path c), rather than a β-hydride elimination (path d),4 leads to the product, HCl and Pd(0). The latter is then reoxidized by Cu(II)/O2/HCl or p-benzoquinone/HCl. Despite deep investigations, precise mechanistic details are still uncertain, and remain a matter of debate.3a
C bond into H–Pd. A halide-mediated reductive elimination (path c), rather than a β-hydride elimination (path d),4 leads to the product, HCl and Pd(0). The latter is then reoxidized by Cu(II)/O2/HCl or p-benzoquinone/HCl. Despite deep investigations, precise mechanistic details are still uncertain, and remain a matter of debate.3a
 | ||
| Scheme 1 Mechanism of the Wacker reaction. | ||
For processes based on the combination of different parameters, the proposition of a mechanism and the selection of the active species for the determination of the rate law may be difficult. The use of mass spectrometry is more appropriate for the study of complex phenomena. Electrospray Ionization Mass Spectrometry (ESI-MS) is well adapted for the observation of short-lived molecules in protonated, deprotonated or cationic forms issued from catalyzed reactions, and for the proposition of mechanisms.5 For examples, the mechanism of the Heck,6a and Baylis–Hillman,6b reactions have been studied by on-line monitoring and several intermediates have been detected and characterized by ESI-MS and ESI-MS/MS. Dynamic and time-dependent processes were also observed using such technique. These original works were followed by other studies on a variety of catalytic transformations.7 We report herein our investigations on the Wacker reaction with ESI-MS, using benzoquinone as terminal oxidant (Scheme 2), and discuss the nature of the catalytic species.
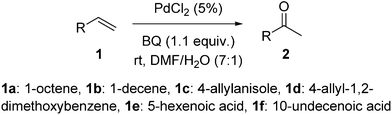 | ||
| Scheme 2 Reactions studied by ESI-MS. | ||
Results and discussion
ESI-MS experiments were first performed in positive ion mode. A mixture of PdCl2 and BQ in DMF/H2O was analyzed (Fig. S1, see supplementary information†). When octene (1a) or decene (1b) was added to the above mixture, new clusters were not observed. Since ionization efficiency by ESI-MS is dependent on the molecules polarity, more polar substrates such as 4-allylanisole (1c) and 4-allyl-1,2-dimethoxybenzene (1d) were tested. New clusters were detected but with low intensity, and the interpretation of the results proved to be difficult (Fig. S2†). We then turned our attention to the use of ESI-MS in the negative ion mode.In comparison to the previous results, the analysis of the PdCl2, BQ, DMF/H2O mixture in the negative ion mode led to a clean spectrum (Fig. 1), and showed three clusters easily attributed to [PdCl3]−, [Pd2Cl5]−, [Pd3Cl7]−, the structures of which were confirmed by High Resolution Mass Spectrometry (HRMS) analysis. Such species correspond to the Cl− adducts of the neutral mononuclear, dinuclear and trinuclear complexes PdCl2, Pd2Cl4, and Pd3Cl6.
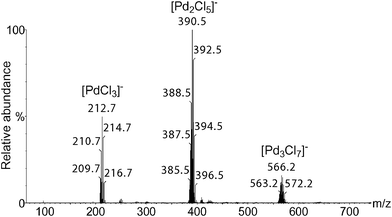 | ||
| Fig. 1 ESI(−)-MS of a mixture of a DMF/water solution of PdCl2 and BQ. Conditions: PdCl2 (0.05 mmol), BQ (1.1 mmol), DMF (1.75 mL), H2O (0.25 mL). | ||
We have subsequently selected substrates which are easily deprotonated such as 5-hexenoic acid (1e) and 10-undecenoic acid (1f) in order to increase the intensity of the signals. Fig. 2 shows the ESI(−)-MS of the crude oxidation reaction of 1e and 1f after 10 min.
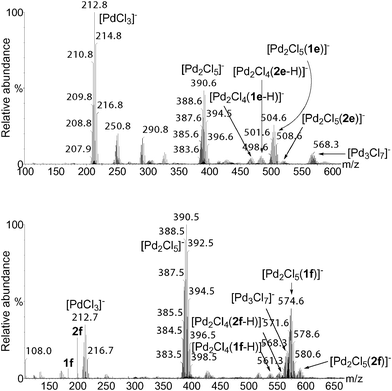 | ||
| Fig. 2 ESI(−)-MS of the reaction solution of the oxidation of 1e (top) and 1f (bottom) after 10 min. | ||
The main feature of the spectra shown in Fig. 2 is that only dinuclear palladium species interacting with 1 and 2 are detectable, no mononuclear species associated with the substrate or the oxidation product being detected.8 Four types of clusters were characterized as [Pd2Cl4(1–H)]−, [Pd2Cl4(2–H)]−, [Pd2Cl5(1)]−, and [Pd2Cl5(2)]− by HRMS. The oxidation of 1f is completed after 2 h as observed by GC analysis. The ESI(−)-MS for such a reaction were recorded after 10, 30, 60 and 120 min (Fig. 3). The four complexes [Pd2Cl4(1–H)]−, [Pd2Cl4(2–H)]−, [Pd2Cl5(1)]−, and [Pd2Cl5(2)]− which were present after 10 min, disappeared progressively, showing that these compounds are associated with transient species.
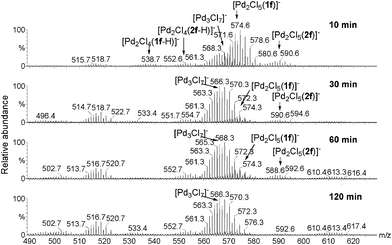 | ||
| Fig. 3 ESI(−)-MS of the reaction solution of the oxidation of 1f over 2 h. | ||
According to ESI(−)-MS/MS experiments, the fragmentations of [Pd2Cl4(1–H)]− and [Pd2Cl4(2–H)]− require an energy (15–20 eV) higher than for [Pd2Cl5(1)]− and [Pd2Cl5(2)]− (6–8 eV). The ESI(−)-MS/MS of the two latter complexes show that the main fragmentation corresponds to the loss of the substrate or the product (Fig. 4 and Fig. S3†). Careful examination of the ESI(−)-MS/MS of [Pd2Cl5(1e)]− and [Pd2Cl5(2e)]− also shows the loss of H2O and HCl, but with a low intensity (Fig. S4†). This is probably due to the presence of signals corresponding to [Pd2Cl4(H2O)(OH)(1e)]− or [Pd2Cl4(H2O)(OH)(2e)]− which overlay with the signals of [Pd2Cl5(1e)]− or [Pd2Cl5(2e)]− respectively. Indeed, both types of clusters have a similar isotopologue distribution (Fig. S5†).
![ESI(−)-MS/MS (6–8 eV) of [Pd2Cl5(1e)]− and [Pd2Cl5(2e)]−.](/image/article/2012/RA/c2ra01204a/c2ra01204a-f4.gif) | ||
| Fig. 4 ESI(−)-MS/MS (6–8 eV) of [Pd2Cl5(1e)]− and [Pd2Cl5(2e)]−. | ||
The ESI(−)-MS/MS of [Pd2Cl4(1–H)]− and [Pd2Cl4(2–H)]− are more complex. The main fragmentation is the loss of PdCl(1–H) or PdCl(2–H) suggesting that substrate and product are strongly coordinated to palladium (Fig. 5 and Fig. S6†). The loss of Cl(1e–H), Cl(2e–H), and Cl(2f–H) is also observed.
![ESI(−)-MS/MS (15–20 eV) of [Pd2Cl4(1e–H)]− and [Pd2Cl4(2e–H)]−.](/image/article/2012/RA/c2ra01204a/c2ra01204a-f5.gif) | ||
| Fig. 5 ESI(−)-MS/MS (15–20 eV) of [Pd2Cl4(1e–H)]− and [Pd2Cl4(2e–H)]−. | ||
We suspected that the dinuclear transient species could be formed due to the bifunctional nature of substrates 1e and 1f. The double bond and the carboxylic function could lead to the coordination of two Pd(II). We therefore pursued our study by ESI-MS using the less functionalised substrates 1a–1d.
Similar results were obtained with such substrates. Indeed, dinuclear species Pd2Cl4(1–H)−, Pd2Cl4(2–H)−, Pd2Cl5(1)−, Pd2Cl5(2)− were also observed, although with lower intensities in certain cases (Fig. S7†). No mononuclear species associated with the substrate or the oxidation product were detected. The ESI(−)-MS/MS of Pd2Cl5(1b–d)− using a low energy of fragmentation showed the loss of 1 (Fig. S8†). The intensities of [Pd2Cl5(1a)]− and Pd2Cl5(2a–d)− were too weak for ESI(−)-MS/MS analysis. The ESI(−)-MS/MS of [Pd2Cl4(1–H)]− and [Pd2Cl4(2–H)]− required higher energy than for Pd2Cl5(1)− and Pd2Cl5(2)− as observed previously, and showed the loss of PdCl(1–H) and PdCl(2–H), as well as the loss of Cl(1–H), or Cl(2–H) in some cases (Fig. S9†).
Pd2Cl5(1)− corresponds to the neutral species Pd2Cl4(1) and is formed during the first step of the catalytic cycle by the coordination of 1 to the dinuclear species Pd2Cl4. The abstraction of chloride atoms by Pd(II) in Pd2Cl4 increases the Lewis acidity at the metal center, and probably makes this species more prompt to react with 1 than the mononuclear complex PdCl2.
The presence of Pd2Cl5(2)− is very surprising, since this species suggests that 2 is coordinated to Pd(II), while the traditional catalytic cycle involves the free ketone 2, its formation coinciding with the liberation of HPdCl or Pd(0) + HCl (Scheme 1). We have studied by ESI(−)-MS the interaction of PdCl2 with 2b, 2c and 2f in DMF/H2O in the presence of BQ under concentrations similar to those used for the oxidations of 1b, 1c and 1f. We did not observe any Pd2Cl5(2)− or Pd2Cl4(2–H)− clusters, even in the case of 2f, while Pd2Cl5(2f)− or Pd2Cl4(2f–H)− are both detected during the oxidation reaction (compare Fig. 2 and Fig. 6). These data suggest that the regeneration of the active Pd species occurs before the decoordination of the product.
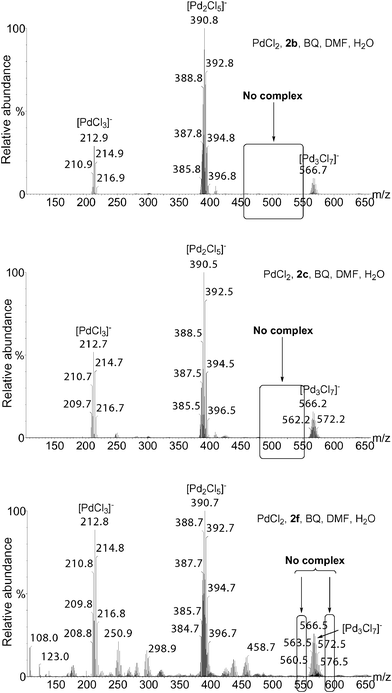 | ||
| Fig. 6 ESI(−)-MS of mixtures of PdCl2 with 2b, 2c or 2f and BQ in DMF/H2O. 2 (1.0 mmol), BQ (1.1 mmol), PdCl2 (0.05 mmol), DMF (1.75 mL) and H2O (0.25 mL). | ||
Pd2Cl4(1–H)− and Pd2Cl4(2–H)− correspond formally to the loss of HCl from Pd2Cl5(1)− and Pd2Cl5(2)−. The ESI(−)-MS/MS of Pd2Cl4(1–H)− and Pd2Cl4(2–H)− have shown that 1 and 2 are strongly coordinated to palladium. Therefore, these clusters could be associated to π-allyl or π-oxo-allyl complexes respectively. The loss of Cl(1–H) and Cl(2–H) in the ESI(−)-MS/MS of Pd2Cl4(1–H)− and Pd2Cl4(2–H)− corresponds to the loss of chlorinated substrates or products formed by reductive elimination from π-allyl-complexes.
Bimetallic species have been discussed in some cases as intermediates in Wacker reactions but no direct proof of their existence was given.9 The Wacker reaction occurs at high [Cl−] and [CuCl2] but is inhibited under high [Cl−] and low [CuCl2].9b Oxgaard and Goddard have shown that at high [Cl−] and [CuCl2], the association of CuCl2 with PdCl2 causes the overall barriers to drop enough to overcome the [Cl−] inhibition.9b Hosokawa et al. disclosed that the reaction of Pd(II) with Cu(II) under O2 gives Pd–Cu heterometallic complexes that catalyze the oxidation of 1-decene into 2-decanone.9c–e The same team also proposed that the formal oxidation state of palladium(II) remains constant throughout the oxidative cyclisation of alkenyl phenols carried out with [(η3-pinene)PdOAc]2 in the presence of Cu(OAc)2 and O2; binuclear RPdH–Cu(OAc)2 and RPdOOH–Cu(OAc)2 species being proposed as catalytic intermediates.10 With Pd(OAc)2, the active species would be a homometallic dinuclear species.11
Bimetallic catalysts are known to present synergistic cooperation between the metal centers. Two metals can share for example the redox work, and therefore, can decrease the activation barriers compared to mononuclear catalysts.12
A simplified mechanism is proposed in Scheme 3. The solubilization of (PdCl2)n leads to dinuclear Pd2Cl4(S)2, which is detected as [Pd2Cl5]− after desolvation and chloride capture. The coordination of 1 affords Pd2Cl4(1)(S) as π and π-allyl complexes in equilibrium. π-Allyl complexes were mentioned in the early studies of the Wacker process by Hafner, but are not usually discussed.1a Such complexes are also proposed as intermediates in the Pd(II)-catalyzed alkenes migration,13 and 1 is indeed isomerized to some extent during the oxidation reaction as observed by GC-MS. Interestingly, a new mechanism, involving dinuclear Pd(II) species, has been recently proposed for the (RCN)2PdCl2-catalyzed E/Z interconversion in alkenes.14 DFT calculations have shown that an alkene can add to a bis-chloro bridged Pd(II) complex to form a monobridged species. The latter facilitates reversible binuclear 1,2-chloropalladation of the alkene through the formation of a six-membered ring adduct.14 A similar mechanism could occur in the Wacker reaction, the hydroxypalladation involving a Pd–alkene unit and an hydroxyl group on the “other” Pd center (Scheme 4).
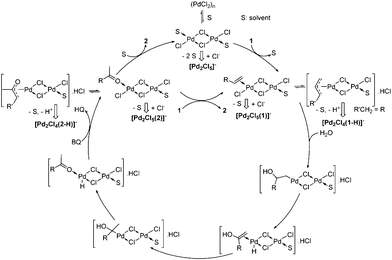 | ||
| Scheme 3 Proposed mechanism. | ||
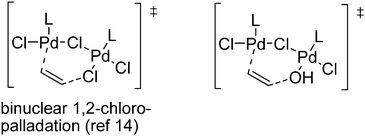 | ||
| Scheme 4 Proposed hydroxypalladation. | ||
Hydroxypalladation would be followed by β-hydride elimination and then insertion of the CC bond into H–Pd. Reductive elimination would lead to the hydride complex [Pd2Cl3(2)(H)(S)]·HCl, which would be rapidly oxidized by BQ to provide Pd2Cl4(2)(S) as π or π-oxo-allyl complexes. Mixed-valence Pd(II)–Pd(0) or Pd(I)–Pd(I) dimers could also be involved. The latter are known to react with O2.15 We did not detect by ESI(−)-MS any complexes showing the coordination of BQ as ligand. Bäckvall has observed π-allyl palladium complexes with BQ as ligand by 1H NMR, but only under specific conditions.16 The catalytic cycle would be ended by the substitution of 2 by 1 or the solvent.
The kinetics of oxidation of 1a monitored by GC revealed a palladium-dependency that was greater than first-order (Fig. 7).
![Dependence of initial rate on [Pd] for the oxidation of 1a. Data are averaged from three reactions.](/image/article/2012/RA/c2ra01204a/c2ra01204a-f7.gif) | ||
| Fig. 7 Dependence of initial rate on [Pd] for the oxidation of 1a. Data are averaged from three reactions. | ||
We have observed by GC that when a mixture of PdCl2 and BQ in DMF/H2O was stirred for 20 h before the addition of 1a, the oxidation rate is slower (Fig. 8). The ESI(−)-MS spectrum of a mixture of PdCl2 and BQ in DMF/H2O stirred for 20 h shows that [Pd2Cl5]− is less present than under standard conditions (compare Fig. 1 and Fig. 9).
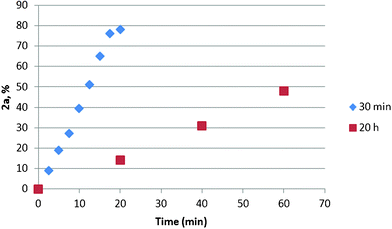 | ||
| Fig. 8 Kinetics of oxidation of 1a. A mixture of PdCl2 (0.05 mmol) and BQ (1.1 mmol) in DMF (1.75 mL)/H2O (0.25 mL) was stirred for 30 min or 20 h before the addition of 1a (1.0 mmol). | ||
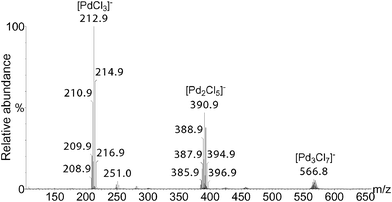 | ||
| Fig. 9 ESI(−)-MS of a mixture of PdCl2 and BQ in DMF/H2O stirred for 20 h. PdCl2 (0.05 mmol), BQ (1.1 mmol), DMF (1.75 mL), H2O (0.25 mL). | ||
We have then analyzed by ESI(−)-MS mixtures of PdCl2 and BQ in DMF/H2O using different amounts of PdCl2 (Fig. 10). The concentration of the polynuclear species increased with the amount of Pd as expected, and among these species [Pd2Cl5]− becomes predominant. The monitoring of the oxidation of 1f by ESI(−)-MS using 0.25% of PdCl2 has shown the presence of a cluster at m/z 398.7 that could be attributed to the mononuclear cluster [PdCl3(1f)]−, which has however a lower abundance than the dinuclear analog [Pd2Cl5(1f)]− (Fig. 11). Careful analysis of ESI(−)-MS performed with 0.5 to 5.0% PdCl2 has shown that [PdCl3(1f)]− is not detected for these higher concentrations of palladium.
![Dependence of ESI(−)-MS of a mixture of PdCl2 + BQ in DMF/H2O on [Pd].](/image/article/2012/RA/c2ra01204a/c2ra01204a-f10.gif) | ||
| Fig. 10 Dependence of ESI(−)-MS of a mixture of PdCl2 + BQ in DMF/H2O on [Pd]. | ||
 | ||
| Fig. 11 ESI(−)-MS of the reaction solution of oxidation of 1f with 0.25% of PdCl2 after 10 min. 1f (20 mmol), PdCl2 (0.05 mmol), BQ (22 mmol), DMF (35 mL), H2O (5 mL). | ||
These results are in accordance with the kinetic experiments and suggest that the dinuclear species Pd2Cl4 is more involved than the mononuclear species PdCl2 under Tsuji–Wacker conditions.
Conclusion
Mechanistic proposals of the Wacker reaction usually involved mononuclear catalytic intermediates. Dinuclear species have been discussed in certain cases. The present study, using ESI-MS, shows that dinuclear palladium complexes are more involved than mononuclear species under the classical conditions which use benzoquinone as terminal oxidant. We propose that the catalytic cycle occurs through the coordination of the substrate to Pd2Cl4, an hydroxypalladation followed by the usual elimination/insertion sequences leading to an hydride Pd complex. This complex would be reoxidized before the decomplexation of the product. We believe these data will provide an anchor point to guide further mechanistic and computational studies.Experimental
General information
Solvents and reagents were used as received. Kinetic experiments were performed by GC using PhNO2 as internal standard. Electrospray ionization mass spectrometry experiments (MS and HRMS) were obtained on a hybrid tandem quadrupole/time-of-flight (Q-TOF) instrument, equipped with a pneumatically assisted electrospray (Z-spray) ion source (Micromass, Manchester, UK). ALPHAGAZ AR2 gas was used for CID. The electrospray potential was set to 3 kV in positive ion mode, and the extraction cone voltage was usually varied between (30–60 V, flow of injection 5 μL min−1). Spectra were typically an average of 20–40 scans. Theoretical isotope patterns calculated with the Isoform program were used to aid assignment. In order to obtain valid exact mass measurement, an external reference or “lock mass” was used to correct for changes in environment or experimental conditions over the course of the analysis. The LockSpray dual electrospray ion source optimizes the co-introduction of analyte and “lock mass” compound directly into the ion source, providing authenticated exact mass measurement in MS modes to within 5 ppm RMS mass accuracy.General procedure for ESI-MS experiments
A round bottom flask was charged with BQ (120 mg, 1.1 mmol), PdCl2 (8.8 mg, 0.05 mmol), DMF (1.75 mL) and H2O (0.25 mL). The mixture was stirred for 30 min, then 1 (1.0 mmol) was added. The reaction mixture was directly injected into the ESI-MS spectrometer at regular intervals of time.ESHRMS data
[PdCl3]−: Cl3Pd: calcd: 210.8100; Found: 210.8102.[Pd2Cl5]−: Cl5Pd2: calcd: 386.6512; Found: 386.6522.
[Pd3Cl7]−: Cl7Pd3: calcd: 562.4924; Found: 562.4918.
[Pd2Cl4(2a–H)]−: C8H15OCl4Pd2: calcd: 478.7947; Found: 478.7946.
[Pd2Cl5(1a)]−: C8H16Cl5Pd2: calcd: 498.7764; Found: 498.7765.
[Pd2Cl4(1b–H)]−: C10H19Cl4Pd2: calcd: 490.8310; Found: 490.8312.
[Pd2Cl4(2b–H)]−: C10H19OCl4Pd2: calcd: 506.8260; Found: 506.8270.
[Pd2Cl5(1b)]−: C10H20Cl5Pd2: calcd: 526.8077; Found: 526.8069.
[Pd2Cl4(1c–H)]−: C10H11OCl4Pd2: calcd: 498.7634; Found: 498.7625.
[Pd2Cl4(2c–H)]−: C10H11O2Cl4Pd2: calcd: 514.7583; Found: 514.7575.
[Pd2Cl5(1c)]−: C10H12OCl5Pd2: calcd: 534.7400; Found: 534.7396.
[Pd2Cl4(1d–H)]−: C11H13O2Cl4Pd2: calcd: 528.7739; Found: 528.7731.
[Pd2Cl5(1d)]−: C11H14O2Cl5Pd2: calcd: 564.7506; Found: 564.7504.
[Pd2Cl4(2d–H)]−: C11H13O3Cl4Pd2: calcd: 544.7688; Found: 544.7701.
[Pd2Cl4(1e–H)]−: C6H9O2Cl4Pd2: calcd: 464.7426; Found: 464.7417.
[Pd2Cl4(2e–H)]−: C6H9O3Cl4Pd2: calcd: 480.7375; Found: 480.7368.
[Pd2Cl5(1e)]−: C6H10O2Cl5Pd2: calcd: 500.7193; Found: 500.7200.
[Pd2Cl5(2e)]−: C6H10O3Cl5Pd2: calcd: 516.7142; Found: 516.7153.
[Pd2Cl4(1f–H)]−: C11H19O2Cl4Pd2: calcd: 534.8209; Found: 534.8212.
[Pd2Cl4(2f–H)]−: C11H19O3Cl4Pd2: calcd: 550.8158; Found: 550.8156.
[Pd2Cl5(1f)]−: C11H20O2Cl5Pd2: calcd: 570.7975; Found: 570.7986.
[Pd2Cl5(2f)]−: C11H20O3Cl5Pd2: calcd: 586.7925; Found: 586.7932.
References
- (a) R. Jira, Angew. Chem., Int. Ed., 2009, 48, 9034 CrossRef CAS; (b) J. Muzart, Tetrahedron, 2007, 63, 7505 CrossRef CAS; (c) J. M. Takacs and X.-t. Jiang, Curr. Org. Chem., 2003, 7, 369 CrossRef CAS.
- (a) J. Tsuji, H. Nagashima and H. Nemoto, Org. Synth., 1984, 62, 9 CAS; (b) J. Tsuji, Synthesis, 1984, 369 CrossRef CAS.
- For a recent review on the mechanism of the Wacker reaction, see: (a) J. A. Keith and P. M. Henry, Angew. Chem., Int. Ed., 2009, 48, 9038 CrossRef CAS. For subsequent studies on the mechanism of the Wacker reaction, see: (b) N. N. Nair, J. Phys. Chem. B, 2011, 115, 2312 CrossRef CAS; (c) A. Comas-Vives, A. Stirling, A. Lledós and G. Ujaque, Chem.–Eur. J., 2010, 16, 8738 CrossRef CAS; (d) B. J. Anderson, J. A. Keith and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 11872 CrossRef CAS.
- J. A. Keith, J. Oxgaard and W. A. Goddard, J. Am. Chem. Soc., 2006, 128, 3132 CrossRef CAS.
- (a) L. S. Santos, Reactive Intermediates: MS Investigations in Solution, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) C. A. Müller, C. Markert, A. M. Teichert and A. Pfaltz, Chem. Commun., 2009, 1607 RSC; (c) D. M. Chisholm and J. S. McIndoe, Dalton Trans., 2008, 3933 RSC; (d) L. S. Santos, Eur. J. Org. Chem., 2008, 235 CrossRef CAS; (e) M. N. Eberlin, Eur. J. Mass Spectrom., 2007, 13, 19 CrossRef CAS; (f) P. Chen, Angew. Chem., Int. Ed., 2003, 42, 2832 CrossRef CAS.
- (a) A. A. Sabino, A. H. L. Machado, C. R. D. Correia and M. N. Eberlin, Angew. Chem., Int. Ed., 2004, 43, 2514 CrossRef CAS; (b) L. S. Santos, C. H. Pavam, W. P. Almeida, F. Coelho and M. N. Eberlin, Angew. Chem., Int. Ed., 2004, 43, 4330 CrossRef CAS.
- (a) K. L. Vikse, Z. Ahmadi, C. C. Manning, D. A. Harrington and J. Scott McIndoe, Angew. Chem., Int. Ed., 2011, 50, 8304 CAS; (b) T. Regiani, V. G. Santos, M. N. Godoi, B. G. Vaz, M. N. Eberlin and F. Coelho, Chem. Commun., 2011, 47, 6593 RSC; (c) F. F. D. Oliveira, M. R. dos Santos, P. M. Lalli, E. M. Schmidt, P. Bakuzis, A. A. M. Lapis, A. L. Monteiro, M. N. Eberlin and B. A. D. Neto, J. Org. Chem., 2011, 76, 10140 CrossRef CAS; (d) M. A. Henderson, J. Luo, A. Oliver and J. S. McIndoe, Organometallics, 2011, 30, 5471 CrossRef CAS; (e) M. W. Alachraf, P. P. Handayani, M. R. M. Hüttl, C. Grondal, D. Enders and W. Schrader, Org. Biomol. Chem., 2011, 9, 1047 RSC; (f) K. L. Vikse, M. A. Henderson, A. G. Oliver and J. S. McIndoe, Chem. Commun., 2010, 46, 7412 RSC; (g) M. A. Schade, J. E. Fleckenstein, P. Knochel and K. Koszinowski, J. Org. Chem., 2010, 75, 6848 CrossRef CAS; (h) T. A. Fernandes, B. Gontijo Vaz, M. N. Eberlin, A. J. M. da Silva and P. R. R. Costa, J. Org. Chem., 2010, 75, 7085 CrossRef CAS; (i) C. H. Beierlein, B. Breit, R. A. Paz Schmidt and D. A. Plattner, Organometallics, 2010, 29, 2521 CrossRef CAS; (j) G. W. Amarante, H. M. S. Milagre, B. G. Vaz, B. R. V. Ferreira, M. N. Eberlin and F. Coelho, J. Org. Chem., 2009, 74, 3031 CrossRef CAS; (k) G. W. Amarante, M. Benassi, H. M. S. Milagre, A. A. C. Braga, F. Maseras, M. N. Eberlin and F. Coelho, Chem.–Eur. J., 2009, 15, 12460 CrossRef CAS; (l) E. Thiery, D. Harakat, J. Le Bras and J. Muzart, Organometallics, 2008, 27, 3996 CrossRef CAS; (m) L. S. Santos, G. B. Rosso, R. A. Pilli and M. N. Eberlin, J. Org. Chem., 2007, 72, 5809 CrossRef CAS; (n) C. Markert, M. Neuburger, K. Kulicke, M. Meuwly and A. Pfaltz, Angew. Chem., Int. Ed., 2007, 46, 5892 CrossRef CAS; (o) C. A. Marquez, F. Fabbretti and J. O. Metzger, Angew. Chem., Int. Ed., 2007, 46, 6915 CrossRef CAS; (p) P.-A. Enquist, P. Nilsson, P. Sjöberg and M. Larhed, J. Org. Chem., 2006, 71, 8779 CrossRef CAS; (q) C. Marquez and J. O. Metzger, Chem. Commun., 2006, 1539 RSC; (r) H. Guo, R. Qian, Y. Liao, S. Ma and Y. Guo, J. Am. Chem. Soc., 2005, 127, 13060 CrossRef CAS; (s) A. Pla-Quintana and A. Roglans, Arkivoc, 2005, 9, 51 Search PubMed.
- Trinuclear species associated with 1 or 2 are not observed. Due to the low intensity of the signal associated with [Pd3Cl7]−, it is however difficult to affirm that they are not involved in the catalytic cycle.
- (a) Y. Kawamura, Y. Kawano, T. Matsuda, Y. Ishitobi and T. Hosokawa, J. Org. Chem., 2009, 74, 3048 CrossRef CAS; (b) J. A. Keith, R. J. Nielsen, J. Oxgaard and W. A. Goddard, J. Am. Chem. Soc., 2007, 129, 12342 CrossRef CAS; (c) T. Hosokawa, T. Nomura and S.-I. Murahashi, J. Organomet. Chem., 1998, 551, 387 CrossRef CAS; (d) T. Hosokawa, T. Nomura and S.-I. Murahashi, J. Organomet. Chem., 1998, 566, 293 CrossRef CAS; (e) T. Hosokawa, M. Takano and S.-I. Murahashi, J. Am. Chem. Soc., 1996, 118, 3990 CrossRef CAS.
- T. Hosokawa and S.-I. Murahashi, Acc. Chem. Res., 1990, 23, 49 CrossRef CAS.
- D. D. Kragten, R. A. van Santen and J. J. Lerou, J. Phys. Chem. A, 1999, 103, 80 CrossRef CAS.
- D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard and T. Ritter, J. Am. Chem. Soc., 2010, 132, 14092 CrossRef CAS.
- D. R. Chrisope, P. Beak and W. H. Saunders, J. Am. Chem. Soc., 1988, 110, 230 CrossRef CAS.
- E. H. P. Tan, G. C. Lloyd-Jones, J. N. Harvey, A. J. J. Lennox and B. M. Mills, Angew. Chem., Int. Ed., 2011, 50, 9602 CrossRef CAS.
- (a) R. Huacuja, D. J. Graham, C. M. Fafard, C.-H. Chen, B. M. Foxman, D. E. Herbert, G. Alliger, C. M. Thomas and O. V. Ozerov, J. Am. Chem. Soc., 2011, 133, 3820 CrossRef CAS; (b) V. Durà-Vilà, D. M. P. Mingos, R. Vilar, A. J. P. White and D. J. Williams, Chem. Commun., 2000, 1525 RSC.
- Interaction of BQ with (π-allyl)PdX complexes was observed by 1H NMR for X = BF4, but was not detected in the presence of strong binding chloride ligands (X = Cl), see J.-E. Bäckvall and A. Gogoll, Tetrahedron Lett., 1988, 29, 2243 CrossRef.
Footnote |
| † Electronic supplementary information available. See DOI: 10.1039/c2ra01204a |
| This journal is © The Royal Society of Chemistry 2012 |