Computational design by evolving folds and assemblies over the alphabet in L- and D-α-amino acids†
Punam
Ghosh
,
Ameeq ul
Mushtaq
and
Susheel
Durani
*
Bioorganic Laboratory, Department of Chemistry, Indian Institute of Technology Bombay, Mumbai, 400 076, India. E-mail: sdurani@iitb.ac.in
First published on 7th March 2012
Abstract
A step by step search of polypeptide structure, first for folds over L and D structures, then assemblies over the folds, and finally sequences over the assemblies, by inverse application of the protein alphabet in side chains, is introduced as a practical algorithm for the evolutionary design of biologically-inspired supramolecules tailored to a desired size, shape, and function. With stereochemistry and symmetry as powerful considerations for the design of, respectively, folds and assemblies, two independent octapeptides are accomplished, over the alphabet of L- and D-α-amino acids, as remarkably small Shiga-toxin mimics.
Nature’s algorithm for protein design was extended in scope while it was simplified in execution by harnessing residue stereochemistry as a design variable.1 The variable not only diversifies folds but also simplifies design, being the director of folds more resolute than the protein alphabet in side chains. The codes in the alphabet of α-amino acid structures, defining protein folds and functions, remain obscure in their working basis.2–4 Thus the design of the codes for polypeptides to adopt desired folds and manifest desired functions remains a challenge. The designs have been approached beneficially with empirical functions for interactions of side chains, which are implemented for inverse optimization of folds as proteins using suitable computational algorithms.5–8 The progress has been impressive but remains restricted in the scope of possible targets to only what is feasible with the natural alphabet. Stereochemistry has been shown to offer promise for minimizing and diversifying structures, as well as for simplifying the design of protein-like structures.1 In one simplification, computation was recruited for the possibility of step-by-step design, first of folds over L and D structures and then of sequences by inverse application of the protein alphabet in side chains.9,10 Demonstrating the approach, two unrelated mimics of Shiga toxin11 are reported in this study as structures dramatically smaller, with eight-residue building blocks, than the natural structure, which has building blocks sixty-eight residues in length. Evolutionary design of biologically inspired supramolecules is demonstrated with application of stereochemistry and symmetry as powerful considerations for implementing the design of first polypeptide folds and then assemblies over the folds.
With α-helix and β-sheet building blocks of poly-L structure, protein structures are constrained stereochemically to be all-α, all-β, or mixed-α,β folds. The all-β Trp-zip of 16 residues,12 the all-α villin headpiece of 35 residues,13 and the mixed-α,β BBA of 21 residues14 exemplify the smallest poly-L proteins possible; stereochemistry limits folds and thus proteins. We have proposed diversifying folds, and thus proteins, stereochemically—bracelet-, boat-, canoe-, and cup-shaped proteins were presented as illustrations of the folds and the proteins possible over L- and D-α-amino acids as the design alphabet.15–18 Small folds of L and D structures may be diverse enough structures for the design of stable assemblies, even though the folds are unstable in isolation. Exploring this possibility, we implemented searches, first through stereochemical options in L- and D-α-amino acids and then chemical options in side chains, and reported a stereochemically-bent hairpin as a boat-shaped dimer of C2 symmetry.19 Extending the approach we now report a computational algorithm for stepwise design, involving rounds of searches through options of first stereochemistry, then assembly, and finally chemical sequences over side chains. We prove the evolutionary algorithm with the successful design of Ia, Ib, IIa, and IIb as remarkably small Shiga-toxin mimics (Fig. 1).
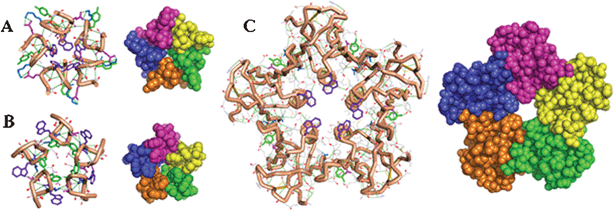 | ||
| Fig. 1 Wire frame and space filling models comparing homopentamers of octapeptide Ia (A) and IIa (B) with Shiga toxin (C) highlighting the clustered aromatics at the cores of the assemblies. | ||
Ac-Leu8NHMe was modeled for the 256 stereoisomers possible with each residue L or D in structure. The structures were ordered to ten random folds over every stereoisomer. The ordering was implemented with molecular dynamics (MD) using GROMACS.20 A Gromos-96 force field21 modified for accommodation of D residues and for preservation of geometry in peptide groups and α carbons of polypeptide structure was implemented in a simulated-annealing algorithm, which was implemented under vacuum.22 Proteins self assemble in cyclic and translational symmetry operations as homomers of closed stoichiometric structure or polymers of open stoichiometric structure; the consideration of symmetry has been discussed extensively for importance in organization and evolution of biomolecular hierarchies and value in the design of materials involving self assembly.23–25 Aiming to test the design algorithm we targeted C5-symmetric Shiga toxin as a model (Fig. 1). Using SymmDock26,27 each one of 2560 vacuum-annealed (VA) folds generated over stereoisomers of Ac-Leu8NHMe was screened for fitness as a building block for self assembly of a homo-pentamer structure. The eighteen highest scoring VA folds shown in Fig. 2 were shortlisted for further study. The selected monomers were targeted for a further round of selection involving evaluation of conformational stability. This evaluation was implemented with MD. Each fold was submitted to MD in a solvent bath of water for 1 ns period. The folds harvested from the MD trajectories at 10 ps intervals were clustered to 0.15 nm RMSD cutoff over Cα coordinates. On this basis, each VA fold was found to relax to multiple clusters ranging from 1 to 11 (†ESI, Table S1). The implication is that the folds are greatly varied in conformational stability as monomers; the most stable fold could be expected to relax to the least number of conformational clusters. The most populous cluster in each starting VA structure was treated, in its central member, as the most stable fold for the covalent structure of the VA fold. The most stable fold in each structure was treated as solvent-relaxed (SR) folds, making it the second generation of fold in the evolutionary design. Many SR folds are noted (†ESI, Table S1) to have relaxed to more main chain hydrogen bonds than the corresponding VR folds, making them potentially better design targets. SR folds were tested for reassembly with SymmDock, and the scores shown in Fig. 2 are, interestingly, improvements over the scores of the VA folds. The highest scoring SR folds (#1487 and #2161) with the highest of main chain hydrogen bonds were shortlisted for further design. The shortlisted folds are 1D2D3D4L5L6L7L8D (Fold I) and 1L2L3D4L5L6D7D8D (Fold II) as the stereochemical structures and 1R2R3R4L5L6R7L8R (Fold I) and 1L2L3R4L5L6R7R8R (Fold II) as the conformational constructs considering the half, left (L) or right (R), of the Ramachandran φ, ψ map (†ESI, Fig. S1 and Table S2) to which the position-specific residues are locked. All residues, with the notable exception of residue 7 in fold I, are noted to have the φ, ψs that represent the stereochemically preferred option for the residue L or D in the structure. Clearly, each fold is stereospecific for its sequences over the alphabet in L and D structure. Thus locked to the desired conformation due to the stereochemical structure in the sequence, each fold could be a conformational construct more robust than a homochiral fold poly-L or poly-D in structure.
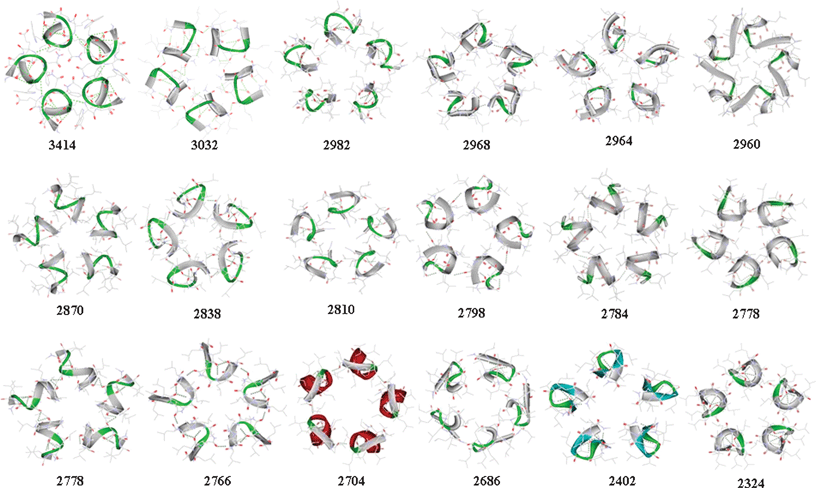 | ||
| Fig. 2 Pentamer assemblies over solvent-relaxed octapeptide folds showing the eighteen highest scoring monomers in diminishing SymmDock scores. | ||
Inverse design was implemented against the folds in assembly; thus the folds and the assemblies were targeted for locking in situ with the effects of sequence. Sequence optimizations, involving inverse application of the protein alphabet in side chains, were implemented with in-house software IDeAS.9 The software applies side chains from a database of natural L rotamers28,29 and their symmetry transforms, which are generated for the residues of D structure. Coordinated application of identical rotamers in C5-symmetry-related positions over the building-block monomers was implemented in rounds of dead end elimination30 followed by Monte Carlo search cycles.31 Hydrogen-bond energy,32 solvent-accessibility-based hydrophobic effect,33 and Lennard-Jones energy34 were applied as the search functions.35–37 Rotamer selection for sequence optimization was constrained to allow π–π, cation–π, hydrogen bonding, salt bridging, and hydrophobic-pair interactions, intramolecularly in the folds and intermolecularly in the assemblies. Specific interactions in favor of folding and assembly of the desired structures are listed in Table 1. Tryptophan and phenylalanine were placed as hubs around the symmetry axis of the pentamer structure in mimicry of the arrangement in Shiga toxin and in tryptophan-zipper- and phenylalanine-zipper-pentamer structures.38,39 The sequence variants a and b have 2Glu or 2Gln as alternatives in fold I and 8Ala or 8Arg as alternatives in fold II. The charge change from neutral Ia and IIa to cationic Ib and IIb (†ESI, Fig. S2) is planned for examining possible effects on the solubility of monomers and the stability of homomers.
| Intramolecular interactions | Intermolecular interactions |
|---|---|
| Ia: Ac-DSDEDTLWLLLKLYDV-NH2 | |
| Ib: Ac-DSDQDTLWLLLKLYDV-NH2 | |
| H-Bond: 1SNH-5LCO, 5LNH-1SCO, 3TNH-1SCO, 4WNH-1SCO, 4WNH-2E/2QCO, 4WNH-7YCO, 7YNH-5LCO | H-Bond: 6KNH-2ECO |
| Cation–π: 6K-7Y | Salt bridge: 2E-6K |
| Hydrophobic interaction: 4W-5L | π–π: 4W-4W |
| Hydrophobic interaction: 8V-8V | |
| IIa: Ac-LTLKDWLFLNDEDVDA-NH2 | |
| IIb: Ac-LTLKDWLFLNDEDVDR-NH2 | |
| H-Bond: 2KNH-8A/8RCO, 3WNH-8A/8RCO, 5NNH-2KCO, 2KNH-5NCO, 8A/8RNH-3WCO | H-Bond: 6ENH-2KCO |
| π–π: 3W-4F | Salt bridge: 6E-2K |
| Cation–π: 3W-8R | π–π: 4F-4F; 3W-4F |
| Hydrophobic interaction: 4F-7V | |
The folds and assemblies were assessed for stability in water with MD, which was run for 50 ns duration. The structures harvested from MD trajectories at 10 ps intervals were clustered to 0.15 nm RMSD cutoff over Cα atoms. The folds were found to diverge to clusters of discrete conformation ranging from 5 to 198 in number (†ESI, Table S3 and Fig. S3). The most populous cluster recovered from MD was found to encompass ranging from 13 to 97% of all the folds sampled during MD; accordingly, SR folds vary considerably in conformational stability. Likewise, homomers were found to cluster in ranging from 450 to 898 discrete structures (†ESI, Table S3 and Fig. S4), and the most populous clusters were noted to encompass ∼5% of the structures sampled over cationic Ib and IIb and >20% over neutral Ia and IIa. It is possible that the charge change from a to b will affect the folds and the assemblies in stability.
Synthesis40 by solid-phase method gave the structures that manifested the requisite peaks in MALDI-MS (†ESI, Fig. S5) and expected resonances in 1H NMR spectra (†ESI, Fig. S6). The charged variants Ib and IIb were freely soluble in water while the neutral variants Ia and IIa necessitated methanol as co-solvent for full solubility. Proton chemical shifts were assigned for Ib with 2D NMR41,42 (†ESI, Table S4 and Fig. S7, S8). J values were extracted directly from 1D spectra (†ESI Table S5). Conformational modeling was implemented with CYANA43 using inter-proton distances, which were calibrated over NOE volumes, as the distance constraints (†ESI, Table S6) for structure solution. Structure calculation was implemented over the constraints. The modeled lowest energy folds are shown in Fig. 3 superposed over the central member of the largest cluster for the fold recovered from MD. The folds are in RMSD of 0.52 nm over their Cα coordinates.
![Superposition of the five lowest energy CYANA-derived folds in Ib [mutual Cα RMSD = 0.52 Å] (Panel A), of central members of the five most populous clusters of the fold recovered from MD [mutual Cα RMSD = 1.1 Å] (Panel B), and of the lowest energy CYANA model, the central member of the most populous MD cluster, and the design generated computationally [mutual Cα RMSD = 4.2 Å] (Panel C).](/image/article/2012/RA/c2ra01012g/c2ra01012g-f3.gif) | ||
| Fig. 3 Superposition of the five lowest energy CYANA-derived folds in Ib [mutual Cα RMSD = 0.52 Å] (Panel A), of central members of the five most populous clusters of the fold recovered from MD [mutual Cα RMSD = 1.1 Å] (Panel B), and of the lowest energy CYANA model, the central member of the most populous MD cluster, and the design generated computationally [mutual Cα RMSD = 4.2 Å] (Panel C). | ||
Self assembly was evaluated by monitoring CD, fluorescence, and NMR as a function of concentration, which was varied 1000 fold from 5 μM to 5 mM. CD studies were undertaken in 20% methanol–water mixture for neutral Ia and IIa and in pure water for charged Ib and IIb (Fig. 4). Ellipticities were found to change in the 5 to 100 μM concentration range of the peptides and noted to achieve a practical constancy in the 100 to 250 μM concentration range (†ESI, Fig. S9). Coupled exciton with a minimum at ∼200 nm and a maximum at ∼220 nm appears in Ia and Ib and evidences possible interaction of the Trp residue intermolecularly in a π–π interaction. A weak isosbestic appears with all the peptides and the change of ellipticity with concentration saturates to a negative value at 230 nm. Ellipticity at this wavelength manifests a sigmoidal relationship with concentration of the peptides (inset in Fig. 4); the midpoint of the transition appears at ∼50 μM in Ia, IIa, and IIb and at a slightly higher concentration in Ib.
 | ||
| Fig. 4 CD spectra of neutral and charged octapeptides as a function of concentration. Ellipticity at λ 230 nm as a function of concentration (inset). | ||
All peptides were noted to manifest quenching of fluorescence (Fig. 5, Panel A) and a reduction of fluorescence anisotropy with increase of concentration (Fig. 5, Panel B). The observed anisotropy change is unusual for increase of mass with self assembly. Energy transfer is implied from quenching self assembly (Fig. 5, Panel A). This may be involved in the anomalous depolarization change with increase of peptide concentration. Both fluorescence quenching and anisotropy changes are noted to be much larger for neutral Ia and IIa than for charged Ib and IIb; however, the concentration threshold for self assembly appears to be in a comparable 50–100 μM range for all the structures. The changes of CD and fluorescence reach a practically flat baseline with concentration for all the structures; this conforms to possible ordering of the structures as closed stoichiometric complexes of point-group symmetry. NMR spectra were evaluated for effects of diluting the samples tenfold from 5.0 to 0.5 mM concentration (†ESI, Fig. S10). Charged Ib and IIb are by and large unaffected by dilution while neutral Ia and IIa manifest appreciable chemical shift changes in some of the peptide-NH resonances. The structures may be prone to self assembly in millimolar concentration regimes as possible higher-order aggregates.
 | ||
| Fig. 5 Fluorescence emission spectra of neutral and charged peptides as a function of concentration (Panel A). Fluorescence intensity (inset Panel A) and anisotropy (Panel B) as a function of concentration. | ||
An attempt was made to characterize self assembly with dynamic light scattering (DLS) and atomic force microscopy (AFM). According to DLS, IIa and IIb manifest, with increasing concentration, particles that are progressively higher in hydrodynamic radius ranging from low ∼2 nm, to intermediate ∼5–20 nm, and even higher values are indicated (†ESI, Fig. S12). With AFM, spherical particles of 38.0 ± 4.6 and 43.7 ± 2.3 nm diameter are observed with Ib and IIb in high resolution measurements, while similar particles of much larger size, more copiously with neutral Ia and IIa than with charged Ib and IIb, are observed in low resolution measurements (†ESI, Fig. S13). The results with DLS represent the detection limit for the technique44–46 in respect of both concentration regime of the experiment and the size of the particles implied, which appear to be close to the molecular dimensions for monomer fold and pentamer assembly. The results with AFM manifest the smallest particles that could pack up to 50 monomer folds in self assembly. A categorical proof for pentamer self assembly necessitates more rigorous DLS experiments or alternative detection methods, while the nature of larger structures detected with AFM remains unclear. However, the observation of particles of closed symmetry progressively larger in size implies a hierarchical process that may involve the partners interacting in a point-group symmetric operation. Conformity with our design strategy, the application of point-group symmetry operation as the approach for self assembly, is noteworthy.
A practical method for the evolutionary design of biologically-inspired supramolecules over heterochiral polypeptide structure has been proposed. The method involves computer-assisted searches of first folds over L and D structures, then assemblies over the folds, and finally sequences over the assemblies in amino acid side chains. The approach was shown to furnish folds and assemblies over peptides that had simple enough structures to be easily accessed by chemical synthesis. Following the proposed diversification of folds involving the application of stereochemistry as a design variable,1 a practical approach for evolutionary selection, at the level of stereochemical and chemical options of sequence, of desired folds and assemblies has been presented as a technological leap in molecular and material design.
Acknowledgements
We acknowledge DST (SR/S1/OC-74/2008), Government of India, for financial support, and IIT Bombay for providing supercomputing facility “Corona”. We also acknowledge the national NMR facility at TIFR, Mumbai, for the use of NMR instruments. Dipanwita De gave useful suggestions for recording of fluorescence anisotropy. The AFM data were recorded at the SPM Facility, Department of Physics, IIT Bombay.References
- S. Durani, Acc. Chem. Res., 2008, 41, 1301 CrossRef CAS.
- R. L. Baldwin, J. Biol. Chem., 2003, 278, 17581 CrossRef CAS.
- G. D. Rose, P. J. Fleming, J. R. Banavar and A. Maritan, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16623 CrossRef CAS.
- K. A. Dill, S. B. Ozkan, M. S. Shell and T. R. Weikl, Annu. Rev. Biophys., 2008, 37, 289 CrossRef CAS.
- G. Butterfoss and B. Kuhlman, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 49 CrossRef CAS.
- R. Das and D. Baker, Annu. Rev. Biochem., 2008, 77, 363 CrossRef CAS.
- S. Lippow and B. Tidor, Curr. Opin. Biotechnol., 2007, 18, 1 CrossRef.
- S. Park, S. Yang and J. Saven, Curr. Opin. Struct. Biol., 2004, 14, 487 CrossRef CAS.
- R. Ranbhor, A. Tendulkar, A. Kumar, V. Ramakrishnan, K. Patel, K. R. Srivastava and S. Durani, submitted, 2011.
- R. Ranbhor, A. Kumar, K. Patel, V. Ramakrishnan, P. Gupta, B. Goyal, K. R. Srivastava and S. Durani, submitted, 2011.
- P. E. Stein, A. Boodhoo, G. J. Tyrrel, J. L. Brunton and R. J. Read, Nature, 1992, 355, 748 CrossRef CAS.
- A. G. Cochran, N. J. Skelton and M. A. Starovasnik, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 5578 CrossRef CAS.
- M. G. Oakley and P. S. Kim, Biochemistry, 1998, 37, 12603 CrossRef CAS.
- M. D. Struthers, R. P. Cheng and B. Imperiali, Science, 1996, 271, 342 CAS.
- S. Rana, B. Kundu and S. Durani, Chem. Commun., 2004, 2462 RSC.
- S. Rana, B. Kundu and S. Durani, Chem. Commun., 2005, 207 RSC.
- S. Rana, B. Kundu and S. Durani, Bioorg. Med. Chem., 2007, 15, 3874 CrossRef CAS.
- S. Rana, B. Kundu and S. Durani, Biopolymers, 2007, 87, 231 CrossRef CAS.
- P. Ghosh, P. Dutta, D. Pednekar and S. Durani, submitted, 2011.
- W. F. van Gunsteren, S. R. Billeter, A. A. Eising, P. H. Hunenberger, P. Kruger, A. E. Mark, W. R. P. Scott and I. G. Tironi, Biomolecular Simulation: The GROMOS96 manual and user guide, Hochschulverlag AG an der ETH Zurich, Zurich, Switzerland, 1996 Search PubMed.
- E. Lindahl, B. Hess and D. van der Spoel, J. Mol. Model., 2001, 7, 306 CAS.
- V. Ramakrishnan, R. Ranbhor and S. Durani, Biopolymers, 2005, 78, 96 CrossRef CAS.
- T. L. Blundell and N. Srinivasan, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14243 CrossRef CAS.
- J. E. Padilla, C. Colovos and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 2217 CrossRef CAS.
- T. O. Yeates and J. E. Padilla, Curr. Opin. Struct. Biol., 2002, 12, 464 CrossRef CAS.
- D. S. Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Nucleic Acids Res., 2005, 33, W363 CrossRef.
- D. S. Duhovny, Y. Inbar, R. Nussinov and H. J. Wolfson, Proteins: Struct., Funct., Bioinf., 2005, 60, 224 CrossRef.
- R. L. Dunbrack, Curr. Opin. Struct. Biol., 2002, 12, 431 CrossRef CAS.
- S. C. Lovell, J. M. Word, J. S. Richardson and D. C. Richardson, Proteins: Struct., Funct., Genet., 2000, 40, 389 CrossRef CAS.
- R. F. Goldstein, Biophys. J., 1994, 66, 1335 CrossRef CAS.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087 CrossRef CAS.
- D. B. Gordon, S. A. Marshall and S. L. Mayo, Curr. Opin. Struct. Biol., 1999, 9, 509 CrossRef CAS.
- B. I. Dahiyat and S. L. Mayo, Protein Sci., 1996, 5, 895 CrossRef CAS.
- D. N. Bolon, J. S. Marcus, S. A. Ross and S. L. Mayo, J. Mol. Biol., 2003, 329, 611 CrossRef CAS.
- A. G. Street and S. L. Mayo, Structure, 1999, 7, R105 CrossRef CAS.
- B. Kuhlman, G. Dantas, G. C. Ireton, G. Varani, B. L. Stoddard and D. Baker, Science, 2003, 302, 1364 CrossRef CAS.
- S. M. Lippow and B. Tidor, Curr. Opin. Biotechnol., 2007, 18, 1 CrossRef.
- J. Liu, W. Yong, Y. Deng, N. R. Kallenbach and M. Lu, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16156 CrossRef CAS.
- J. Liu, Q. Zheng, Y. Deng, N. R. Kallenbach and M. Lu, J. Mol. Biol., 2006, 361, 168 CrossRef CAS.
- W. C. Chan and P. D. White, Fmoc Solid Phase Peptide Synthesis: A Practical Approach, IRL Press, Oxford, UK, 1989 Search PubMed.
- D. G. Davis and A. Bax, J. Am. Chem. Soc., 1985, 107, 2821 CrossRef.
- A. Kumar, R. R. Ernst and K. Wuthrich, Biochem. Biophys. Res. Commun., 1980, 95, 1 CrossRef CAS.
- P. Guntert, C. Mumenthaler and K. Wuthrich, J. Mol. Biol., 1997, 273, 283 CrossRef CAS.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330 CrossRef CAS.
- X. Wang, J. F. Graveland-Bikker, C. G. De Kruif and G. T. Robillard, Protein Sci., 2004, 13, 810 CrossRef CAS.
- A. V. Shkumatov, S. Chinnathambi, E. Mandelkow and D. I. Svergun, Proteins: Struct., Funct., Bioinf., 2011, 79, 2122 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Sequence design and validation, synthesis, mass spectra, NMR data, circular dichroism, fluorescence and computational data. See DOI: 10.1039/c2ra01012g |
| This journal is © The Royal Society of Chemistry 2012 |