Oxidizing Morita–Baylis–Hillman adducts towards vicinal tricarbonyl compounds†
Marília S.
Santos
and
Fernando
Coelho
*
University of Campinas, Laboratory of Natural Products and Drugs Synthesis, PO Box 6154-13083-970–Campinas, São Paulo, Brazil. E-mail: coelho@iqm.unicamp.br; Fax: +55 19 35213023; Tel: 55 19 35213085
First published on 20th January 2012
Abstract
We disclose herein a simple and direct method to synthesize vicinal tricarbonyl compounds (VTC) from Morita–Baylis–Hillman (MBH) adducts. The method is based on two sequential oxidation steps which provide VTC in moderate to good yields. This is the first report of the synthesis of vicinal tricarbonyl compounds from MBH adducts.
Introduction
The presence of at least three contiguous carbonyl groups in a given molecule defines what we ordinarily call vicinal tricarbonyl compounds (VTC). This system is characterized by having a highly electrophilic central carbonyl, which makes it very reactive towards different nucleophiles and leads to the synthetic versatility of this structural arrangement. These compounds can be used as building blocks for the synthesis of heterocycles having unusual substitution patterns1 and biologically active natural products, such as antibiotics,2,3 and compounds with immunosuppressive and vasodilator activities.3,4 Vicinal tricarbonyl compounds are one of the most powerful acceptors for nucleophilic residues and therefore can act as potent inhibitors of hydrolytic enzymes, such as proteases and elastases (Fig. 1).5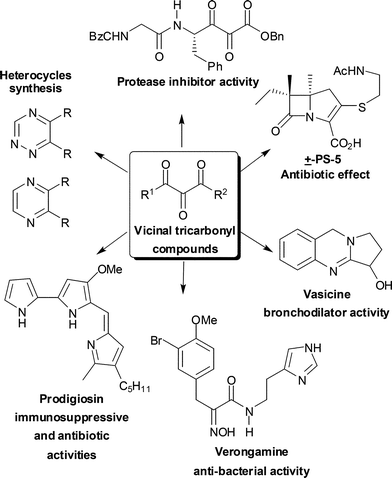 | ||
| Fig. 1 VTC as building blocks for organic synthesis. | ||
The synthetic versatility of VTC has stimulated efforts toward their preparation,6 and important contributions have been made by Wasserman7 in this research field. Basically, VTC are synthesized via suitable functionalization of 1,3-dicarbonyl compounds, which places a new functional group between the two carbonyls for further oxidation via DMSO/singlet oxygen,7a Swern,8 Dess–Martin,9 SeO2,7c and ozonolysis5a,b (Fig. 2). Selective double insertion of aryl isocyanide mediated by samarium(II) diiodide or diol oxidation with 2,2,6,6-tetramethylpiperidine-1-oxyl (4-BzO-TEMPO) combined with NaBrO2·3H2O have also been employed.10
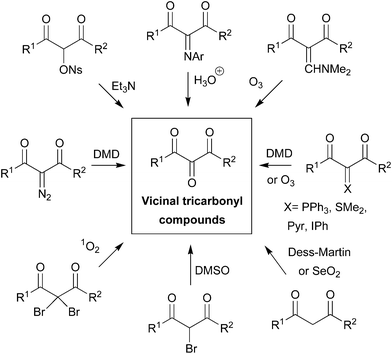 | ||
| Fig. 2 VTC from 1,3-dicarbonyl compounds.‡ | ||
The Morita–Baylis–Hillman (MBH) reaction is a sustainable organocatalyzed chemical transformation which provides highly functionalized small molecules.11 MBH adducts have been efficiently used for the preparation of natural products and drugs.12
Recently we13 and Douteau14 have reported the successful ozonolysis of the double bond of Morita–Baylis–Hillman adducts. It is worth noting that the ozonolysis reaction can be troublesome for MBH adducts, especially for those derived from aromatic aldehydes, since they are prone to extensive oxidative degradation.15 We have successfully circumvented the overoxidation issues and used this strategy as a key step for the synthesis of natural products, like styryl lactones.16
The ozonolysis of the MBH adducts provides α-keto-β-hydroxyesters, and we envisaged that these compounds could function as substrates for a simple new oxidation step leading to VTC in a direct and simple manner (Scheme 1).
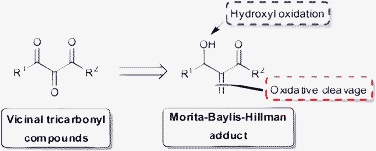 | ||
| Scheme 1 Ease of access to VTC from MBH adducts. | ||
The biological and chemical relevance of VTC justify the development of alternative methods to their synthesis. Thus, we describe herein a new approach to the preparation of VTC from MBH adducts. This two step oxidation sequence provides the required compounds in good to excellent overall yield.
Results and discussion
First we prepared some MBH adducts using the methodology developed by us some years ago.17 The adducts were obtained in good yield and the results are summarized in Table 1.|
|
|||
|---|---|---|---|
| Entry | R1 | R2, adducta | Yield (%)b,c |
| a Typically, a mixture of aldehyde, DABCO and an excess of acrylate was placed into an ultrasound bath for some hours. b Yields refer to isolated and purified products. c Spectral data (1H- and 13C NMR) are compatible for all compounds (see ESI†). | |||
| 1 | Phenyl | R2 = OMe, 1 | 85 |
| 2 | Phenyl | R2 = OEt, 2 | 83 |
| 3 | 4-MeO-phenyl | R2 = OMe, 3 | 71 |
| 4 | 4-Isopropylphenyl | R2 = OMe, 4 | 66 |
| 5 | 4-O2N-phenyl | R2 = OMe, 5 | 90 |
| 6 | Phenyl | R2 = Me, 6 | 62 |
| 7 | 3,4,5-Trimethoxyphenyl | R2 = OMe, 7 | 71 |
| 8 | 4-tButylphenyl | R2 = OMe, 8 | 77 |
| 9 | 3-Chlorophenyl | R2 = OMe, 9 | 80 |
| 10 | 4-Bromophenyl | R2 = OMe, 10 | 75 |
| 11 | 4-Isopropylphenyl | R2 = Ot–Bu, 11 | 52 |
| 12 | n-Propyl | R2 = OMe, 12 | 95 |
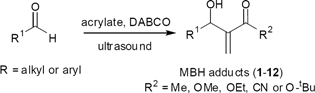
Searching for the best experimental sequence, we used MBH adduct 1 as a model. Compound 1 was allowed to react with ozone at −78 °C in methanol to give a α-keto-β-hydroxyester in 70% yield. The handling of this compound proved to be very difficult. All attempts to purify it by silica gel column chromatography failed and extensive product degradation was observed. We therefore reversed the sequence and began by oxidizing the secondary hydroxyl group of the MBH adduct.
Several alternatives are available to perform the oxidation of an allylic alcohol to an α,β-unsaturated carbonyl compound; however in the case of MBH adduct this is not a trivial exercise.18 Initially we decided to test IBX (2-iodoxybenzoic acid).19 This hypervalent iodine reagent has become reagent of choice due to its several advantages (low toxicity, easy to handle, zero toxic waste and good tolerance to moisture). Besides, IBX has already been used in several chemical transformations involving MBH adducts.20 Adduct 1 was therefore treated with IBX (3 equiv.) in refluxing ethyl acetate to provide the α-methylene-β-ketoester 13 in 92% yield. While trying to optimize this reaction so as to decrease the amount of IBX, we tested some other experimental conditions. The results are summarized in Table 2.
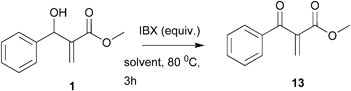
These results show that no significant decrease on the reaction yield is observed when oxidation was performed in acetonitrile; in addition with this solvent it was possible to reduce the amount of IBX in 50% (entry 5). This was selected therefore as the standard conditions for the further oxidations. The results are summarized in Table 3.
|
|
|||
|---|---|---|---|
| Entry | MBH adduct | α-Methylene-β-keto carbonyl compounds | Yield (%) |
| a AcOEt as solvent and 3.0 equiv of IBX. | |||
| 1 | 1 | 13, R1= Phenyl; R2 = OMe | 90 |
| 2 | 2 | 14, R1 = Phenyl; R2= OEt | > 95 |
| 3 | 3 | 15, R1= 4-MeO-phenyl; R2= OMe | 94 |
| 4 | 4 | 16, R1 = 4-Isopropylphenyl; R2 = OMe | > 95 |
| 5 | 5 | 17, R1 = 4-O2N-phenyl; R2= OMe | 90 |
| 6 | 6 | 18 a, R1= Phenyl; R2 = Me | 93 |
| 7 | 7 | 19, R1= 3,4,5-Trimethoxyphenyl; R2= OMe | > 95 |
| 8 | 8 | 20, R1 = tButylphenyl; R2 = OMe | > 95 |
| 9 | 9 | 21, R1 = 3-Chlorophenyl; R2 = OMe | 98 |
| 10 | 10 | 22, R1 = 4-Bromophenyl; R2 = OMe | 95 |
| 11 | 11 | 23, R1 = 4-Isopropylphenyl; R2 = OtBu | > 95 |
| 12 | 12 | 24 a, R1 = n-Propyl; R2 = OMe | 73 |
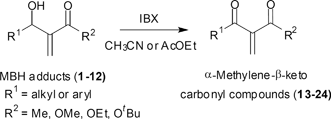
In all cases it was possible to obtain the corresponding α-methylene-β-ketocarbonyl compounds in good yields. In most cases the compounds have high purity and no chromatographic purification was needed. The spectroscopic data (1H NMR) for all compounds show basically the disappearance of the singlet ranging from 4.5 to 5.7 ppm atributted to the carbinolic hydrogen. By other hand, a new carbonyl absorption appeared in the 13C NMR spectra ranging from 191 to 199 ppm.
With the requisite α-methylene-β-keto carbonyl compounds in our hands, we undertook the last step of our synthetic sequence. Pleasingly, when the dicarbonyl compound 13 was treated with a current of ozone (0.3 to 0.4% of ozone in a flow of dry oxygen) in dichloromethane at −78 °C, for 30 min, the expected VTC 25 was obtained in 78%. In most cases no chromatographic purification was needed (Table 4, entry 1). Then ozonolysis of the remaining dicarbonyl compounds (14–24) was performed by using either dichloromethane or methanol as the solvents. As it can be seen in Table 4 product yields are somewhat better in the former solvent (see entries 1, 3 and 7). In this way several VTCs were prepared in moderate to good yields.
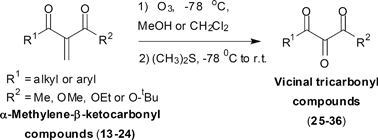
| Entry | α-Methylene-β-keto carbonyl compounds | Vicinal tricarbonyl compounds | Yield (%)e |
|---|---|---|---|
| a Ozonolysis was carried out in CH2Cl2 for 12–30 min, at −78 °C. b Ozonolysis was performed in MeOH for 10–50 min., at −78 °C. c Acetone was used as solvent. d Benzoic acid derivative was detected, this could indicate that oxidative cleavage has occurred on the formed VTC.21 e Yields refer to isolated and purified products. f Yield in MeOH. | |||
| 1 | 13 | 25 a | 78 (57)f |
| 2 | 14 | 26 a | > 95 |
| 3 | 15 | 27 b | 77 (22)f |
| 4 | 16 | 28 a | 76 |
| 5 | 17 | 29 c | < 10 |
| 6 | 18 | 30 b , d | ND |
| 7 | 19 | 31 | 68 (10)f |
| 8 | 20 | 32 a | > 95 |
| 9 | 21 | 33 a | 65 |
| 10 | 22 | 34 a | 75 |
| 11 | 23 | 35 a | 61 |
| 12 | 24 | 36 a | 22 |
The ozonolysis works efficiently allowing the synthesis of several VTC. The products are stable and can be purified by column chromatography, when necessary. We observes a significant decrease in yield (10–20%) when the aromatic ring is substituted by strong electron-donating or electron-withdrawing groups. Trying to circumvent these limitations, the reactions were performed in the most diluted conditions in dichloromethane. A net improvement was observed for some cases (see, entries 3 and 7).
MBH adducts prepared from others acrylates (methylvinylketone and acrolein) were also tested. IBX oxidations worked efficiently for the adduct obtained with methyl vinyl ketone (entry 6, Table 3). Both oxidation steps failed when the MBH adduct obtained from acrolein was used as substrate.
The central carbonyl of these tricarbonyl compounds is highly electrophilic and is prone to nucleophilic addition. Even poor electrophiles like water can lead to the formation of a hydrate. The spectra (1H and 13C NMR) of all VTC prepared in this work show clearly the mixture tricarbonyl + hydrate, as expected (Fig. 3).22
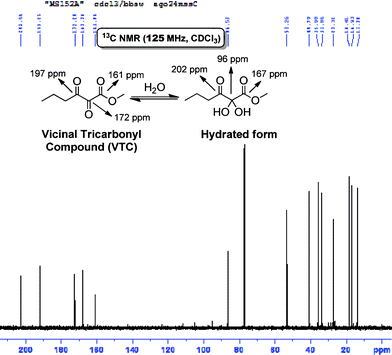 | ||
| Fig. 3 13C NMR (125 MHz, CDCl3) spectrum showing the mixture vicinal tricarbonyl compound + its hydrated form in equal parts. | ||
The 13C NMR (125 MHz, CDCl3) shows five signals attributed to the VTC (3 signals for the tricarbonyl + 2 signals for the hydrate) and a signal at 96 ppm attributed to the central carbon of the hydrate (Fig. 3).
Conclusions
We describe a facile and direct method to prepare VTC from MBH adducts. The method works efficiently for several MBH adducts having ester groups, but reduced yields were observed for cyano or methylketone substituents.As far as we know this is the first report of the preparation of VTC from MBH adducts as substrates. These adducts can be easily synthesized from aldehyde, which renders this method as one of the simplest ways of accessing VTC with low environmental impact since the MBH reaction has no chemical residue. IBX oxidation provides a recyclable byproduct (IBA, o-iodosobenzoic acid), while ozonolysis furnishes formaldehyde, which is more environmentally benign than phosphorus derivatives.3–5
Acknowledgements
MSS thanks Fapesp for a grant. FC thanks Fapesp and CNPq for financial support.References
- (a) V. Nair and A. Deepthi, Tetrahedron Lett., 2006, 47, 2037 Search PubMed; (b) R. M. Adlington, J. E. Baldwin, D. Catterick and G. J. Pritchard, J. Chem. Soc., Perkin Trans. 1, 2001, 668 RSC.
- (a) N. R. Williamson, P. C. Fineran, T. Gristwood, S. R. Chawrai, F. J. Leeper and G. P. Salmond, Future Microbiol., 2007, 6, 605 Search PubMed; (b) J. W. Bennett and R. Bentley, Adv. Appl. Microbiol., 2000, 47, 1 Search PubMed.
- (a) H. H. Wasserman and G. -H. Huo, Tetrahedron Lett., 1991, 32, 7131 CrossRef CAS; (b) H. H. J. Parr and C. M. Baldino, Curr. Opinion Drugs Discovery Dev., 2006, 6, 670 Search PubMed.
- (a) H. H. Wasserman and W. T. A. Han, Tetrahedron Lett., 1984, 25, 3747 CrossRef CAS; (b) H. H. Wasserman and J. Wang, J. Org. Chem., 1998, 63, 5581 Search PubMed.
- (a) H. H. Wasserman, D. S. Ennis, P. L. Power, M. J. Ross and B. Gomes, J. Org. Chem., 1993, 58, 4785 CrossRef CAS; (b) H. H. Wasserman, J. -H. Chen and M. Xia, J. Am. Chem. Soc., 1999, 121, 1401 CrossRef CAS; (c) H. H. Wasserman, J. -H. Chen and M. Xia, Helv. Chim. Acta, 2000, 83, 2607 Search PubMed.
- (a) For a comprehensive review about vicinal tricarbonyl compounds, see: M. B. Rubin and R. Gleiter, Chem. Rev., 2000, 100, 1121 Search PubMed; (b) S. Goswami, A. C. Maity, H.-K. Fun and S. Chantrapromma, Eur. J. Org. Chem., 2009, 1417 CrossRef CAS.
- (a) H. H. Wasserman and J. Parr, Acc. Chem. Res., 2004, 37, 687 CrossRef CAS; (b) M. R. Mahran, W. M. Abdou, M. M. Sidky and H. Wamhoff, Synthesis, 1987, 506 CrossRef; (c) F. Dayer, H. L. Dao, H. Gold, H. R. Gowal and H. Dahn, Helv. Chim. Acta, 1974, 57, 2201 CrossRef CAS.
- (a) T. K. Jones, S. G. Mills, R. A. Reamer, D. Askin, R. Desmond, R. P. Volante and I. Shinkai, J. Am. Chem. Soc., 1989, 111, 1157 CrossRef CAS; (b) T. K. Jones, R. A. Reamer, R. Desmond and S. G. Mills, J. Am. Chem. Soc., 1990, 112, 2998 CrossRef CAS.
- (a) P. K. Somers, T. J. Wandless and S. L. Schreiber, J. Am. Chem. Soc., 1990, 112, 8045 Search PubMed; (b) M. J. Batchelor, R. G. Gillespie, J. M. Golec and C. J. R. Hedgecock, Tetrahedron Lett., 1993, 34, 167 Search PubMed.
- (a) M. Murakami, H. Masuda, T. Kawano, H. Nakamura and Y. Ito, J. Org. Chem., 1991, 56, 1 CrossRef CAS; (b) T. Inokuchi, P. Liu and S. Torii, Chem. Lett., 1994, 1411 CAS.
- (a) M. Shi, F. -J Wang, M. -X. Zhao, Y. Wei, in The Chemistry of the Morita–Baylis–Hillman Reaction; RSC Publishing: Cambrigde, UK, 2011 Search PubMed; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS; (c) R. O. M. A. de Souza and L. S. M. Miranda, Mini-Rev. Org. Chem., 2010, 7, 212 Search PubMed; (d) V. Singh and S. Batra, Tetrahedron, 2008, 64, 4511 CrossRef CAS; (e) D. Basavaiah, K. V. Rao and R. Reddy, Chem. Soc. Rev., 2007, 36, 1581 RSC; (f) W. P. Almeida and F. Coelho, Quim. Nova, 2001, 23, 98 Search PubMed.
- (a) For some examples related to the use of MBH adducts in the total synthesis of natural products and drugs and mechanistic studies, see: G. W. Amarante, M. Cavallaro and F. Coelho, Tetrahedron Lett., 2010, 51, 2597 Search PubMed; (b) G. W. Amarante, M. Benassi, R. N. Pascoal, M. N. Eberlin and F. Coelho, Tetrahedron, 2010, 66, 4370 Search PubMed; (c) G. P. D. Silveira and F. Coelho, Tetrahedron Lett., 2005, 46, 6477 Search PubMed; (d) L. J. Reddy, J. F. Fournier, B. V. S. Reddy and E. J. Corey, Org. Lett., 2005, 7, 2699 CrossRef CAS; (e) W. P. Almeida and F. Coelho, Tetrahedron Lett., 2003, 44, 937 CrossRef CAS; (f) M. A. Feltrin and W. P. Almeida, Synth. Commun., 2003, 33, 1141 CrossRef CAS; (g) P. J. Dunn, J. F. Fournier, M. L. Hughes, P. M. Searle and A. S. Wood, Org. Process Res. Dev., 2003, 7, 244 Search PubMed; (h) R. C. Rossi and F. Coelho, Tetrahedron Lett., 2002, 42, 2797 Search PubMed; (i) C. R. Mateus and F. Coelho, J. Braz. Chem. Soc., 2005, 16, 386 Search PubMed; (j) Y. Iwabuchi, M. Furukawa, T. Esumi and S. Hatakeyama, Chem. Commun., 2001, 2030 RSC; (k) A. Masunari, E. Ishida, G. Trazzi, W. P. Almeida and F. Coelho, Synth. Commun., 2001, 31, 2127 Search PubMed; (l) G. W. Amarante, M. Benassi, H. S. M. Milagre, A. A. C. Braga, F. Maseras, M. N. Eberlin and F. Coelho, Chem. Eur. J., 2009, 15, 12460 CrossRef CAS; (m) J. A. McCauley, K. Nagasawa, P. A. Lander, S. G. Mischke, M. A. Semones and Y. Kishi, J. Am. Chem. Soc., 1998, 120, 7647 CrossRef CAS.
- C. A. M. Abella, P. Rezende, M. F. L. de Souza and F. Coelho, Tetrahedron Lett., 2005, 46, 6495 CrossRef CAS.
- A. Doutheau, M. Freeza, L. Soulère and Y. Queneau, Tetrahedron Lett., 2008, 49, 145 Search PubMed.
- (a) P. R. R. Costa, S. Pinheiro and C. C. Lopes, Tetrahedron Lett., 1985, 26, 4155 Search PubMed; (b) Y. Yoshida, S. Ichikawa, Y. Shinozuka, M. Satoh, K. Mohri and K. Isobe, Heterocycles, 2005, 65, 1481 Search PubMed; (c) R. B. Woodward, M. P. Cava, W. D. Ollis, A. Hunger, H. U. Daeniker and K. Schenker, J. Am. Chem. Soc., 1954, 76, 4749 CrossRef CAS; (d) V. Prelog, J. Battergay and W. I. Taylor, Helv. Chim. Acta, 1948, 31, 2244 CAS.
- P. H. S. Paioti and F. Coelho, Tetrahedron Lett., 2011, 52, 6180 Search PubMed.
- (a) F. Coelho, W. P. Almeida, D. Veronese, C. R. Mateus, E. C. S. Lopes, R. C. Rossi, G. P. C. Silveira and C. H. Pavam, Tetrahedron, 2002, 58, 7437 CrossRef CAS; (b) W. P. Almeida and F. Coelho, Tetrahedron Lett., 1998, 39, 8609 Search PubMed.
- (a) H. M. R. Hoffmann, A. Gassner and U. Eggert, Chem. Ber., 1991, 124, 2475 CrossRef CAS; (b) N. J. Lawrence, J. P. Crump, A. T. McGown and J. A. Hadfield, Tetrahedron Lett., 2001, 42, 3939 CrossRef CAS.
- (a) V. V. Zhdankin, J. Org. Chem., 2011, 76, 1185 CrossRef CAS; (b) A. Duschek and S. F. Kirsch, Angew. Chem., Int. Ed., 2011, 50, 1254 Search PubMed; (c) V. Satam, A. Harad, R. Rajule and H. Pati, Tetrahedron, 2010, 66, 7659 CrossRef CAS; (d) J. D. More and N. S. Finney, Org. Lett., 2002, 4, 3001 CrossRef CAS.
- (a) L. D. S. Yadav, S. V. Prabhakar and P. Rajesh, Synlett, 2010, 1047 Search PubMed; (b) T. Jia-Neng, L. Haoquan and G. Yanlong, Green Chem., 2010, 12, 1772 RSC; (c) L. D. S. Yadav and A. Chhama, Tetrahedron Lett., 2009, 50, 3801 CrossRef CAS; (d) L. D. S. Yadav and A. Chhama, Tetrahedron Lett., 2009, 50, 715 CrossRef CAS; (e) L. D. S. Yadav, A. Chhama and R. Ankita, Tetrahedron Lett., 2008, 49, 6360 CrossRef CAS; (f) J. S. Yadav, B. V. S. Reddy, A. P. Singh and A. K. Basak, Tetrahedron Lett., 2007, 48, 7546 Search PubMed.
- J. Mecinović, R. B. Hamed and C. J. Schofield, Angew. Chem., Int. Ed., 2009, 48, 2796 CrossRef CAS.
- K. Dei, K. Morino, A. Sudo and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2245 Search PubMed.
Footnotes |
| † Electronic Supplementary Information (ESI) available: Spectral data for all prepared compounds. See DOI: 10.1039/c2ra01267g/ |
| ‡ Adapted from ref. 6a. |
| This journal is © The Royal Society of Chemistry 2012 |