Activation of aliphatic C–H bonds by tetracyanobenzene photosensitization. A time-resolved and steady-state investigation†
Stefano
Protti
ad,
Maurizio
Fagnoni
a,
Sandra
Monti
b,
Julien
Réhault
c,
Olivier
Poizat
d and
Angelo
Albini
*a
aPhotoGreen Lab, Department of Chemistry, University of Pavia, V.le Taramelli 12, 27100, Pavia, Italy. E-mail: angelo.albini@unipv.it; Fax: (+)390382987323; Tel: (+)390382987316
bIstituto per la Sintesi Organica e la Fotoreattività (ISOF-CNR), Via P. Gobetti 101, I-40129, Bologna, Italy
cPhysikalisch-Chemisches Institut, Universität Zürich, Winterthurerstrasse, 190, 8057, Zürich, Switzerland
dLaboratoire de Spectrochimie Infrarouge et Raman-LASIR, (UMR 8516 du CNRS), Université des Sciences et Technologies de Lille, Bat C5, 59655, Villeneuve d'Ascq Cedex, France
First published on 6th January 2012
Abstract
The photochemistry of 1,2,4,5-tetracyanobenzene (TCB) in acetonitrile in the presence of representative aliphatic donors (cyclohexane, 1,4-dioxane and triethylamine) has been investigated by femtosecond (fs) and nanoseconds (ns) flash photolysis as well as steady state irradiation in order to define the kinetic frame for the activation of aliphatic C–H bonds. Unlike in the case of aromatics, no ground state complex (except possibly in the case of triethylamine) or exciplex is formed. The lowest excited singlet is quenched and forms the free radical ions. The efficiency of the process, measured by the yield of TCB˙− (0.84 with cyclohexane, 0.15 with 1,4-dioxane, 0.37 with triethylamine) depends on the rate of return electron transfer, low for a high exothermicity. The chemical reaction following the electron transfer step depends on the properties of the radical cation. Thus, with cyclohexane deprotonation is slow and does not occur measurably on a microsecond scale, while on a longer time scale, the formation of alkyl radicals competes with back electron transfer between the free ions. The deprotonation of amine radical cations is faster and the resulting α-aminoalkyl radicals reduce a further molecule of TCB, causing the accumulation of the radical anion which is indefinitely stable in the absence of oxygen.
Introduction
Photoinduced electron transfer (PET) is one of the most investigated topics in photophysics, particularly when involving aromatic molecules, for which the detection of excited states and transient species is easier. For the same reason, aromatics are often used as non consumed sensitizers in electron transfer photosensitization,1 a method that has been applied to a variety of reactions. Among these, one of the most innovative is the activation of aliphatic C–H bonds (Equ. 1).2 Unique features of such a process are the good efficiency, also in cases with weak R–H donors, and the mild conditions, allowing for better control of the reactions compared to the harsh conditions otherwise required. This is particularly interesting in a field where transition metal catalysis, though rapidly developing, offers at the moment a limited alternative.2l| Sens* + R–H → Sens˙− + R–H˙+ | (1) |
In view of these characteristics, this application has some synthetic appeal. Indeed, although PET studies have mainly focused on mechanistic aspects, a few synthetic applications have recently been reported.3 Requisites of a suitable electron transfer photosensitizer are a highly positive reduction potential of the excited state involved (Sens*) and the lack of competing reactions either from the excited state or from the radical anion Sens˙−, so that it is possible to investigate the chemistry of the radical cation unperturbed. Typical sensitizers that fulfil such conditions are aromatic nitriles, among which 1,2,4,5-tetracyanobenzene (TCB), a powerful oxidant in the excited singlet state,4 which has been frequently used in the investigation of electron transfers (ET), in particular with methylarenes (Ar–Me).5 In that case, exciplex fluorescence was observed and the dependence on the structure of the donor and on the solvent polarity allowed the recognition of the intimate mechanism of PET which may involve the sequential formation of a Contact and a Solvent-Separated Radical Ion Pair (CRIP, SSRIP), or independently solvated Radical Ions (RI, Scheme 1a). Besides photophysical processes, photochemical reactions involving benzylic deprotonation also occurred in polar media (Scheme 1b).6,7
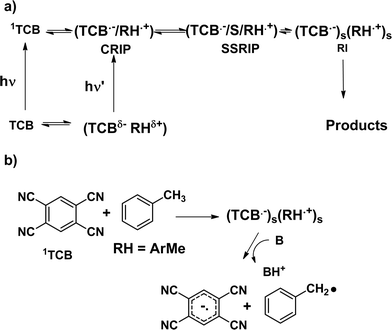 | ||
| Scheme 1 | ||
As mentioned, photosensitized C–H activation is not limited to aromatic donors, but has also been observed in a few instances with aliphatic compounds, including alkanes. In that case, the products obtained were those expected from the trapping of the corresponding alkyl radicals R˙ by the sensitizer or by an electrophilic alkene (see Scheme 2).8–10 In view of the extreme oxidizing character of 1TCB [Ered(1TCB) = Ered(TCB) + Eexc(1TCB) ≈ 3.44 V vs.SCE],7 this may again be rationalized as involving electron transfer from the alkane and deprotonation, although formation of an alkane radical cation in solution is uncommon.11 The rationalization is supported by the fact that the selective deprotonation observed fits well with the calculated preferred fragmentation of the alkane radical cations.9
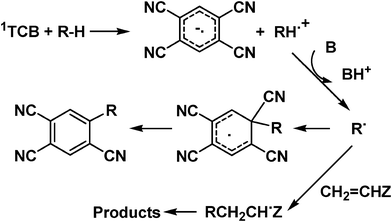 | ||
| Scheme 2 Photoinduced Activation of the C–H bond by TCB. | ||
The TCB photoinduced oxidation of aliphatic compounds has been much less studied than the formation of complexes with aromatics. In view of the synthetic interest of the PET activation of aliphatic C–H bonds, it seemed appropriate that a mechanistic study is carried out.
Experimental section
1,2,4,5-tetracyanobenzene (TCB) and 1,4-diazabicyclo[2.2.2]octane (DABCO) were supplied from Sigma Aldrich and used as received. Triethylamine (TEA), 1,4-dioxane and cyclohexane were commercially available and freshly distilled before use.The femtosecond transient absorption setup has been already described.12 Briefly, it involves a 1 kHz Ti–sapphire laser system based upon a Coherent (MIRA 900D) oscillator and a BM Industries (ALPHA 1000) regenerative amplifier. Pump excitation at 266 nm (<150 fs, 3–15 μJ per pulse, 0.3–1.5 mJ cm−2) was obtained by frequency tripling the fundamental tuned at 800 nm, respectively (0.3 mm BBO crystals). A white light continuum probe pulse was generated by focusing the 800 nm beam in a 1 mm CaF2 plate. The pump–probe polarization configuration was set at the magic angle (54.7°). The probe–pulse was delayed relative to the pump pulse using an optical delay line (Microcontrol Model MT160-250PP driven by an ITL09 controller, precision (1 μm). The overall time resolution (FWHM of the pump–probe intensity cross-correlation) was estimated to be about 300 fs from the two-photon (pump + probe) absorption signal in pure hexane. The time dispersion of the continuum light over the 300–700-nm region of analysis was about 0.8 ps. The transmitted light was sent onto the entrance slit of a 230-mm focal length stigmatic spectrograph equipped with a 150 grooves/mm grating, and analyzed by a charge-coupled device (CCD) optical multichannel analyzer (Princeton Instrument LN/CCD-1340/400-EB detector + ST-138 controller). Sample solutions (OD < 1.0 at 266 nm) were circulated in a flow cell equipped with 0.2 mm thick CaF2 windows and characterized by a 2-mm optical path length. Data were accumulated over 3 min (<180![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pump–probe sequences). Kinetic analyses have been carried out by means of the SPECFIT/32 software (TgK Scientific). Kinetic analyses have been performed by assuming a pulse energy of 0.8 mJ cm−2, thus, for an analyzed volume of 0.255 mm3, 9.6 × 1011 exciting photons have been calculated.
000 pump–probe sequences). Kinetic analyses have been carried out by means of the SPECFIT/32 software (TgK Scientific). Kinetic analyses have been performed by assuming a pulse energy of 0.8 mJ cm−2, thus, for an analyzed volume of 0.255 mm3, 9.6 × 1011 exciting photons have been calculated.
Nano-to-microsecond transient absorption experiments were performed using a nanosecond laser flash photolysis apparatus. Excitation pulses at 266 nm (20 ns, 1 mJ) were provided by a 20-Hz Nd:YAG laser (DIVA II, Thales laser). The probe light was provided by a Xe flash lamp (XBO 150 W/CR OFR, OSRAM). Samples were placed in a quartz cell (10 × 10 mm2 section) at a concentration adjusted to obtain an OD value of 1.0 at 266 nm. The transmitted light was analyzed with a photomultiplier (R1477-06, Hamamatsu) coupled to a digital oscilloscope (TDS 540, Tektronix). Investigations have been performed on nitrogen-purged solutions apart from those used for measuring the effect of quenching by O2.
The UV-Vis absorption spectra were recorded by means of a V-550 Jasco spectrophotometer and the fluorescence spectra by using a LS-55 Perkin Elmer spectrofluorimeter. The accumulation of the TCB˙− species was obtained by irradiation of a freeze–degas–thaw deoxygenated MeCN solution of TCB in the presence of TEA 0.5 M.
Results and discussion
1. Femtosecond to nanosecond time domain
In a mechanistic investigation of the interaction between photoexcited TCB and its aliphatic derivatives, spectroscopic evidence may be sought through the measurement of both the formation and decay of the radical anion TCB˙−, known to absorb strongly in the blue region,13 and flash photolysis is the appropriate technique, as indicated by the successful application to aromatic donors. Accordingly, we report here time-resolved absorption (with both fs and ns resolution) and steady state experiments on the photoexcitation of TCB in MeCN (known from previous studies not to be oxidized under these conditions)13 in the presence of aliphatic donors. The study results, we feel, in a consistent mechanistic picture, as summarized in Schemes 3 and 4 and discussed below.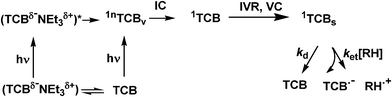 | ||
| Scheme 3 | ||
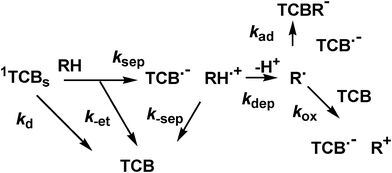 | ||
| Scheme 4 ‘Late’ events in the photochemistry of TCB. | ||
A 7 × 10−4 M solution of TCB in MeCN was excited by means of a 70 fs FWHM laser pulse at 266 nm (corresponding to high-lying singlets, see the TCB absorption spectrum in the ESI†). On the basis of the number of incident photons per laser pulse (see Experimental) a concentration 7 × 10−6 M of excited molecules was calculated. Transient absorption changes showed a complex evolution in the time interval 0.65 ps – 2 ns, as reported in Fig. 1a, and finally disappeared within a few ns.
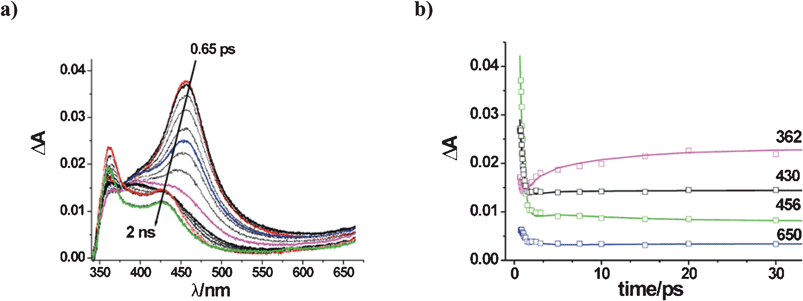 | ||
| Fig. 1 Transients formed upon the excitation of a 7 × 10−4 M solution of TCB in MeCN by means of a 70 fs FWHM laser pulse at 266 nm: (a) Absorbance changes; b) Reproduction of absorbance changes at key wavelengths, as obtained by global kinetic analysis (see text). | ||
The initial spectrum was characterized by an intense band peaking at 456 nm and a weak shoulder around 362 nm, probably truncated on the short-wavelength side due to a negative contribution by fluorescence. The 456 nm band decayed with a subpicosecond time constant (see below) to yield a broad spectrum with a poorly resolved maximum at 400 nm. In turn, this evolved in a few tens of ps, resulting in more defined maxima at 426 and 360 nm, whereas the 400 nm prominence disappeared (further noticeable was a clear rise at 360 nm, after the initial decrease in the subpicosecond domain). From ca. 50 ps up to the ns domain the transient spectrum remained practically unchanged and was therefore assigned to the solvated and vibrationally fully relaxed lowest singlet state, 1TCBs. The complex absorption changes from the initially populated higher singlet to the final relaxed state could be approximated by a multiexponential kinetic model. Global kinetic simulations were performed with the SPECFIT/32 program (see ESI†). Three kinetic components with time constants of 0.44 ps, 1.1 ps and 10 ps were found to reproduce the ΔA(λ,t) reasonably well, up to a 50 ps delay in the whole spectral range explored. The quality of the agreement with the experimental data can be appreciated at 362, 430, 456, 650 nm in Fig. 1b. The two longest time constants were reasonably assigned to relaxation processes in the lowest excited singlet state (1TCBv → 1TCBs), i.e. intramolecular vibrational energy redistribution (IVR) and vibrational cooling (VC). As for the 0.44 ps time constant, two rationalizations have been considered. In the first one, this pertains to the solvent relaxation dynamics (average solvation time of MeCN, <τ>solv = 0.26 ps)14 of the hot lowest singlet 1TCBv, supposed to be already formed by IC from 1nTCBv at this delay. This is supported by the narrowness of the 456 nm band in the 0.65 ps spectrum and agrees with recent evidence for IC processes occurring in < 100 fs with aromatic molecules in MeCN.15 Alternatively, it is this constant that corresponds to IC from the initial state to 1TCBv, as supported by the fact that the 456 ns band decays with no noticeable change in shape, as typical of a depopulation. As for the ‘final’ 1TCBs state, the decay rate could be only roughly estimated from the present transient absorption data as ≈ 4 ns, which compares reasonably well with the fluorescence lifetime in dilute solution (τf = 9 ns), when considering the different conditions of the two experiments. No indication of an interaction of excited TCB with the solvent MeCN was revealed. This is consistent with the lack of photochemical reaction in neat MeCN (although in the presence of moisture a different process sets in) and with the rationalization that an electron-withdrawing substituent such as a cyano group precludes the reaction of alkanes.16
The study was extended to further aliphatic compounds known to quench excited TCB and to react with it. In none of the cases examined did a modification of the TCB ground state absorption spectrum intervene. Upon 266 nm excitation of TCB in MeCN in the presence of 0.5 M cyclohexane, the early transient absorption spectrum was again characterized by two bands at 366 and 457 nm that evolved in a complex way, with the difference that an additional transient species was present at the end of the time window explored (see Fig. 2a). The first part of the evolution (0.65 ps up to 50 ps) was reproduced by application of a three exponential model (see Fig. 2b for the comparison of experimental and calculated absorbance changes at key wavelengths) and resulted in three time constants virtually identical to those observed in neat MeCN (Table 1). Thus, the early events leading to 1TCBs were unaffected.
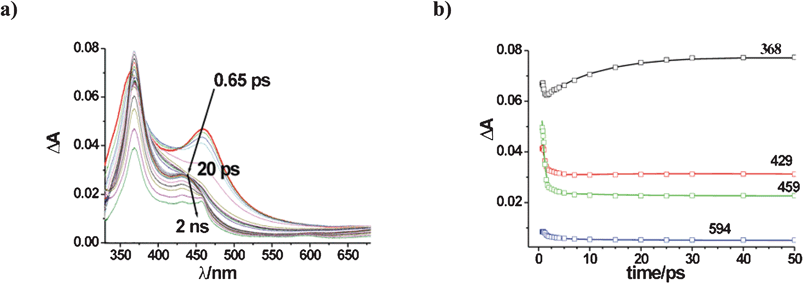 | ||
| Fig. 2 Transients formed upon excitation of a 7 × 10−4 M solution of TCB in MeCN in the presence of 0.5 M cyclohexane. (a) Absorbance changes; b) Reproduction of absorbance changes at key wavelengths, as obtained by global kinetic analysis (see text). | ||
| Additive | k 1 (τ1) | k 2 (τ2) | k 3 (τ3) | k 4 (τ4) |
|---|---|---|---|---|
| None | 2.25 × 1012 s−1 (440 fs) | 9.0 × 1011 s−1 (1.1 ps) | 1.0 × 1011 s−1 (10 ps) | 2.5 × 108 s−1 (4 ns) |
| Cyclohexane 0.5 M | 1.65 × 1012 s−1 (606 fs) | 6 × 1011 s−1 (1.7 ps) | 8.7 × 1010 s−1 (11 ps) | 5.7 × 108 s−1 (1.7 ns) |
| Dioxane 0.3 M | 2.13 × 1012 s−1 (470 fs) | 1.94 × 1011 s−1 (5.2 ps) | 1.15 × 1010 s−1 (87 ps) | |
| TEA 0.2 M | 1.9 × 1012 s−1 (526 fs) | 1.05 × 1012 s−1 (0.95 ps) | 1.2 × 1011 s−1 (8.3 ps) | 5.9 × 109 s−1 (170 ps) |
As for the spectral changes from 50 ps to 1.5 ns, describing the evolution of the excited singlet state 1TCBs from the subnanosecond to the nanosecond domain, a global kinetic analysis was performed, assuming that the starting concentration of this excited state is equal to that of the excited molecules at the end of the laser pulse (7.0 × 10−6 M, see Experimental), i.e. all the relaxation events until 50 ns have unitary efficiency (see ESI† for details on the global kinetic analysis). The decay of the excited state was accompanied by the concomitant formation of a long-lived species with absorption maximum at 460 nm, identified as the radical anion TCB˙−, despite some spectral distortion at short wavelengths. An exponential function was suited to describe the time evolution of the spectral changes. The optimized pseudo first order rate constant, k4, for this process resulted (5.7 ± 0.3)×108 s−1 (τ4 = 1.7 ns), a value that fits fairly well with the Stern–Volmer constant from both steady state and lifetime fluorescence quenching measurements, Ksv = 6 M−1. The global kinetic fitting process allowed to extract the individual profile for the anion separated from that of the excited singlet (see Fig. S3, inset, ESI†). In spite of possible distortions below 400 nm, we believe the long wavelength part in the calculated anion spectrum reliably expresses ε(λ) × Φ, thereby allowing the determination of the quantum yield on the basis of the anion molar absorption coefficient (in polyvinyl alcohol) of 13500 M−1cm−1 at 460 nm.17 A remarkably high value of Φ ≈ 0.8 was calculated. Kinetic profiles at selected wavelengths, in the whole time window explored, with the rate constants in Table 1 are shown in Fig. S4, ESI.†
The early course of the reaction in the presence of 0.5 M 1,4-dioxane (see Fig. S5 and S6, ESI†), was similar to that observed with cyclohexane, with the difference that a three exponential model with time constants 0.47 ps, 5.2 ps and 87 ps was sufficient to describe the evolution up to 500 ps, the two intermediate components ≈ 1–10 ps observed in previous case not being separable here (Table 1). 1TCBs was fully quenched in the subnanosecond domain, giving rise to the radical anion TCB˙− with a quantum yield Φ ≈ 0.13, calculated from the absorbance at 458 nm in the transient spectrum at 500 ps .
In the presence of triethylamine 0.2 M, the transient spectrum at 0.7 ps exhibited bands at 365, 462 and 606 nm (Fig. 3a). The last one, not found with the other quenchers, present at a 0.7 ps delay and disappearing within the early relaxation steps, could be attributed to excitation of a weak complex TEA–TCB (see Scheme 3), for which there is no further evidence (no distinct absorption or fluorescence, ESI†). A four time constant analysis (Table 1 and Fig. S7, ESI†) showed conversion to 1TCBs and subsequent quenching leading to TCB˙− with a time constant of ca. 170 ps, as can be appreciated at 459 nm from ca. 20 ps onwards. An approximate value of Φ ≈ 0.3 was calculated for the quantum yield of formation of TCB˙− from the absorbance at 750 ps.
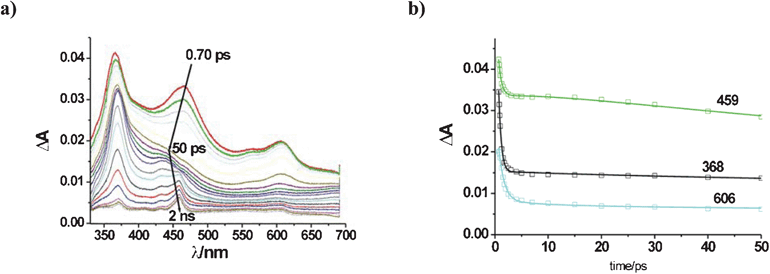 | ||
| Fig. 3 Transients formed upon excitation of a 7 × 10−4 M solution of TCB in MeCN in the presence of 0.2 M triethylamine. (a) Absorbance changes; b) Reproduction of absorbance changes at key wavelengths, as obtained by global kinetic analysis (see text). | ||
2. Nanosecond to microsecond time domain
Further experiments were carried out with nanosecond laser flash photolysis apparatus. Upon laser excitation the absorption spectrum of the radical anion TCB˙− appeared and it was possible to follow its evolution in the μs domain. Representative profiles of the absorbance at λmax (460 nm) are shown in Fig. 4. In the case of cyclohexane, the transient remained almost unchanged at the initial value over the time window explored (up to > 10 microseconds, not shown). As mentioned above, preparative experiments demonstrating the occurrence of radical alkylation processes suggested that alkane radical cations were deprotonated under these conditions8–10 and the resulting radicals attacked the TCB radical anion (see Schemes 2 and 4). Indeed, the addition of alkyl radicals to aromatic radical anions (kad) has been shown to occur efficiently, with a rate within a factor of 3 for compounds of different structure, 1 − 3 × 109 M−1 s−1.18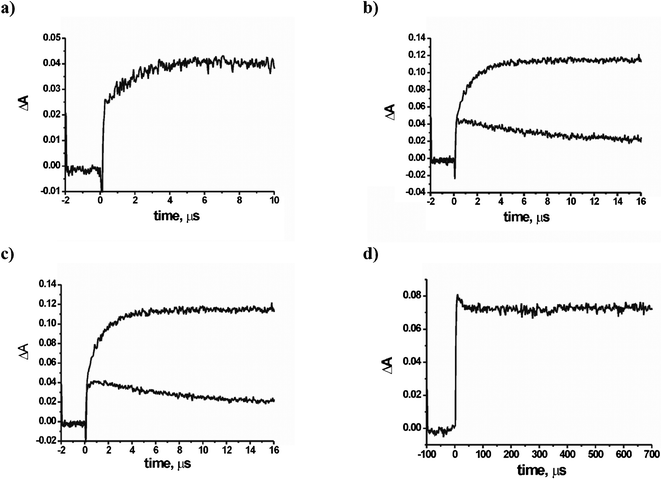 | ||
| Fig. 4 Profile of the absorbance change at 460 nm after excitation of a 7 × 10−4 M TCB solution in nitrogen flushed MeCN with a 20 ns laser pulse at 266 nm in the presence of (a) 0.5 M 1,4-dioxane; (b) 0.2 M triethylamine in oxygen-free (upper trace) and oxygen-equilibrated solutions (lower trace); (c) 0.2 triethylamine (upper trace) and 0.2 M DABCO (lower trace); (d) 0.2 M triethylamine on a longer time window. | ||
The fact that the TCB˙− signal did not diminish in the time window explored was thus due to the inefficient deprotonation of alkanes radical cations, a thermochemically favored process,19 but a slow one in the absence of a base, as it is generally the case for heterolysis of non polar C–H bonds.20,21
In spite of the high quantum yield of the initial electron transfer, radical formation was low, as judged from the inefficient trapping (the TCB alkylation by cyclohexane had Φ = 0.06). In contrast, with dioxane (Fig. 4a) and, more strongly, with triethylamine, the TCB˙− signal grew over the initial value in the microsecond domain. With the amine the ‘slow’ growth was as large as the ‘instantaneous’ part and occurred via a pseudo first order process (Fig. 4b). The fact that the amount of TCB˙− about doubled in some microseconds was explained by the fact that the α-amino radical formed by deprotonation of the amine radical cation (equ. 2, see kox in Scheme 4) was easily oxidized and in particular was oxidized by ground state TCB according to equ. 3.
| Et3N˙+ + Solv (Et3N) → Et2NCH˙Me + SolvH+ (Et3NH+) | (2) |
| Et2NCH˙Me + TCB → Et2NCH+Me + TCB˙− | (3) |
Notice that of the two processes in equ. 2, 3, both involving a transient and a ground state reagent, deprotonation (equ.2) was both a thermochemically allowed process and assisted by the rather concentrated (0.2 M) amine acting as a base. As for the oxidation of the radical by TCB (equ.3) , this was again favored (ΔG = −10 kcal mol−1)22 and occurred at a diffusion controlled rate, but here radical Et2NCH˙Me reacted with less concentrated TCB (7 × 10−4 M). Thus the latter process was the kinetically limiting step and this fitted with the pseudo first-order observed rate (k = 2 × 105 s−1 from Fig. 4b, upper trace).23 Supporting evidence for this rationalization was found in the fact that an amine such as DABCO, for which no α-deprotonation was viable, did not cause the ‘slow’ reduction of TCB (see Fig. 4c, lower trace). Furthermore, repeating the triethylamine experiment under oxygen (Fig. 4b, lower trace) likewise led to the reduction of a single TCB molecule, consistent with preferential trapping of the radical by dissolved oxygen (equ. 4) that hindered the second ET step.
| R˙ + O2 → R–O–O˙ | (4) |
An analogous process took place to some extent with dioxane, in agreement with the fact that the oxidation of the α-oxyalkyl radical, similarly to equ. 3, was non spontaneous (ΔG = + 11 kcal mol−1).22. The relatively slow back oxidation of the TCB radical anion by oxygen (equ. 5), consistent with such a close to thermoneutral (ΔG = −1 kcal mol−1)22 process (see the decay in the microsecond range in Fig. 4b), should also be noted, although traces of oxygen inhibited TCB˙− accumulation, see below).
| TCB˙− + O2 → TCB + O2˙− | (5) |
3. From millisecond domain to persistent species
Another slow process was back electron transfer between the free solvated ions (k-sep, see Scheme 4). Unlike the return electron transfer within the initial encounter complex (k-et), this was a second order process involving two separately solvated transients, too slow to be detected under the conditions of the flash photolysis experiments. Such a process was expected to be even slower under steady state conditions, where the photon flux impinging was many orders of magnitude smaller.24 At any rate, the two processes, k-sep and kad introduced a limit to the attainable concentration of RH˙+ or R˙, unless TCB˙− is consumed or, better, recycled by oxidation to TCB. Stretching the time window to an even longer time scale, radical reactions come into play. Preparative studies showed that the alkyl and α-oxyalkyl radicals were trapped by TCB˙− (kr estimated 109 M−1 s−1, see above)20 or by electrophilic alkenes (these were efficient traps competing with TCB˙−; indeed although the rate constant was lower, kr estimated 106 M−1 s−1,25 the concentration could be much higher). In this way, the corresponding alkyltricyanobenzenes or long chain aliphatic compounds were formed (see Scheme 2).This was not the case for oxidizable radicals such as α-aminoalkyl radicals. These were scooped away according to equ. 4 and the ensuing hydration of the cation to give aldehydes, through an overall two-electrons oxidation. Under these conditions, alkyl radicals were consumed, while no chemical path was available for TCB˙−. The concentration of the two species, initially the same, became different (although the product of these two quantities was limited by k-sep). As seen in flash experiments, [TCB˙−] remained steady over several hundreds of microseconds (see Fig. 4d) and indeed even further, a yellowing of the samples after >10 flashes reveals the accumulation of this species. The steady state irradiation of TCB in the presence of triethylamine was thus carried out and, as it appears in Fig. 5, a 100% reduction to the radical anion was obtained under these conditions. TCB˙− was indefinitely persistent in a deoxygenated solution and was quantitatively oxidized back to the starting material upon oxygen readmission. An analogous accumulation of the radical anion has been previously reported for the case of dicyanoanthracene and dicyanonaphthalene.26
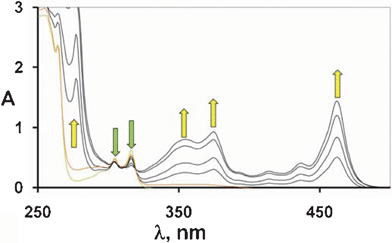 | ||
| Fig. 5 Accumulation of the radical anion of TCB by irradiation of a freeze–degas–thaw deoxygenated MeCN solution in the presence of triethylamine. A 100% conversion into radical anion TCB˙− occurred (yield calculated on the basis of the molar absorption coefficient of TCB*−, see ref. 17). | ||
Conclusion
The TCB photosensitized generation of aliphatic radicals by oxidation-induced bond cleavage (equ. 6) has been studied in different time intervals.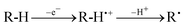 | (6) |
A detailed account of the stepwise process occurring over a time scale >10 orders of magnitude long has been obtained, from the initially formed, high-lying singlet states of the nitrile to the stable end products, passing through the subpicosecond relaxation processes and the kinetically limited reactions of free ions. Contrary to aromatic donors, aliphatic donors form neither ground state complexes (except, possibly, triethylamine) nor exciplexes. Thus, the efficiency of the chemical process with respect to the unproductive physical decay depends on a series of competing processes at various stages, the role of which has been quantified. The main conclusion is that with alkanes the free radical ions are efficiently formed because return electron transfer in the radical ion pair is slower than separation in the ns scale, but then deprotonation of the radical cation is too slow to make this an efficient source of alkyl radicals (in the ms scale and above). On the contrary, with aliphatic amines, the formation of the free radical ions is less efficient, but the (amine assisted) deprotonation is faster, although it does not lead to alkylation but to reduction of a further TCB molecule. This information may help in devising systems suitable for syntheses based on the direct use of alkanes, e.g. for the formation of a C–C bond, provided that deprotonation is made faster and the trap used is able also to reoxidize TCB˙−, thus allowing the nitrile to function as non-consumed sensitizer, as it has been demonstrated in the case of electrophilic alkenes.8 Different applications may be likewise appealing, e.g. for photochromic systems for various time scales or the exploitation of charge separation involving apolar compounds such as alkanes. As a sensitizer, TCB features a strongly oxidizing, relatively long-lived singlet able to cause single electron transfer oxidation of such high Eox reagents as alkanes, as well as a rather inert radical anion that minimally interferes.
Acknowledgements
S. P. acknowledges Egide (Fellowship by the French Embassy in Rome and Italian Ministry of Foreign Affairs) and the Ministero dell'Università e della Ricerca (MIUR), Rome (FIRB-Futuro in Ricerca 2008 project RBFR08J78Q) for financial support. Dr. A. Mezzetti is thanked for his precious help.References
- (a) A. G. Griesbeck, N. Hoffmann and K. D. Warzecha, Acc. Chem. Res., 2007, 40, 128 CrossRef CAS; (b) A. Houmam, Chem. Rev., 2008, 108, 2180 CrossRef CAS; (c) P. A. Waske, N. T. Tzvetkov and J. Mattay, ChemInform, 2007, 38 DOI:10.1002/chin.200727213; (d) M. Ohashi, K. Nakatani, H. Maeda and K. Mizuno, Tetrahedron Lett., 2010, 51, 5537 Search PubMed; (e) N. Hoffmann, J. Photochem. Photobiol., C, 2008, 9, 43 CrossRef CAS; (f) D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999 RSC; (g) H. J. Roth, J. Phys. Chem. A, 2003, 107, 3432 CrossRef CAS; (h) U. C. Yoon and P. S. Mariano, Acc. Chem. Res., 2001, 34, 523 CrossRef CAS; (i) A. Albini, M. Fagnoni and M. Mella, Pure Appl. Chem., 2000, 72, 1321 CrossRef CAS.
- (a) P. J. Wagner, M. J. Thomas and A. E. Puchalski, J. Am. Chem. Soc., 1986, 108, 7739 CrossRef CAS; (b) T. M. Bockman, S. M. Hubig and J. K. Kochi, J. Am. Chem. Soc., 1998, 120, 2826 CrossRef CAS; (c) M. Mella, M. Freccero and A. Albini, Tetrahedron, 1996, 52, 5533 CrossRef CAS; (d) M. Mella, M. Freccero and A. Albini, J. Chem. Soc., Chem. Commun., 1995, 41 RSC; (e) R. Popielarz and D. R. Arnold, J. Am. Chem. Soc., 1990, 112, 3068 CrossRef CAS; (f) J. L. Faria, R. A. McClelland and S. Steenken, Chem.–Eur. J., 1998, 4, 1275 CrossRef CAS; (g) E. R. Gaillard and D. G. Whitten, Acc. Chem. Res., 1996, 29, 292 CrossRef CAS; (h) P. Maslak and S. L. Asel, J. Am. Chem. Soc., 1988, 110, 8260 CrossRef CAS; (i) D. Shukla and R. H. Youngand Samir Farid, J. Phys. Chem. A, 2004, 108, 10386 CrossRef CAS; (j) for metal assisted catalysis see e.g. Y. J. Park, J. W. Park and C. H. Jun, Acc. Chem. Res., 2008, 41, 222 Search PubMed and references quoted therein.
- (a) M. Ohashi, K. Nakatani, H. Maeda and K. Mizuno, J. Org. Chem., 2008, 73, 8348 CrossRef CAS; (b) Z.-F. Lu, Y. M. Shen, J.-J. Yue, H.-W. Hu and J.-H. Xu, J. Org. Chem., 2008, 73, 8010 Search PubMed.
- (a) I. R. Gould and S. Farid, Acc. Chem. Res., 1996, 29, 522 CrossRef CAS; (b) N. Mataga, H. Chosrowjan and S. Taniguchi, J. Photochem. Photobiol., C, 2005, 6, 37 CrossRef CAS and references therein.
- (a) B. R. Arnold, J. S. Atherton, S. Farid, J. L. Goodman and I. R. Gould, Photochem. Photobiol., 1997, 65, 15 Search PubMed; (b) B. R. Arnold, A. Euler, P. V. Poliakov and A. W. Schill, J. Phys. Chem. A, 2001, 105, 10404 Search PubMed; (c) B. R. Arnold, D. Noukakis, S. Farid, J. L. Goodman and I. R. Gould, J. Am. Chem. Soc., 1995, 117, 4399 CrossRef CAS; (d) I. R. Gould, D. Noukakis, L. Gomez-Jahn, J. L. Goodman and S. Farid, J. Am. Chem. Soc., 1993, 115, 4405 CrossRef CAS; (e) H. Lemmetyinen, N. T. Tkachenko, A. Efimov and M. Niemi, J. Phys. Chem. C, 2009, 113, 11475 CrossRef CAS; (f) E. Vauthey, J. Photochem. Photobiol., A, 2006, 179, 1–12 Search PubMed; (g) S. Ojima, H. Miyasaka and N. Mataga, J. Phys. Chem., 1990, 94, 4147 CrossRef CAS; (h) S. V. Feskov, V. N. Ionkin, A. I. Ivanov, H. Hagemann and E. Vauthey, J. Phys. Chem. A, 2008, 112, 594 Search PubMed; (i) I. R. Gould and S. Farid, J. Phys. Chem. B, 2007, 111, 6782 Search PubMed; (j) J. Zhou, C. Zhong, T. M. Francis and C. L. Braun, J. Phys. Chem. A, 2003, 107, 8319 Search PubMed; (k) J. Dresner, J. Prochorow and W. Ode, J. Phys. Chem., 1989, 93, 671 CrossRef CAS; (l) T. Asahi and N. Mataga, J. Phys. Chem., 1989, 93, 6575 CrossRef CAS; (m) N. Mataga, Pure Appl. Chem., 1984, 56, 1255 CrossRef CAS.
- (a) V. Dichiarante, M. Fagnoni and A. Albini, in Modern Arylation Methods (Ed. L. Ackermann) Wiley-Weinheim, 2009, 513–535 Search PubMed; (b) Y. Ito, H. Nakabayashi, S. Ohba and H. Hosomi, Tetrahedron, 2000, 56, 7139 Search PubMed; (c) M. Bellanova, M. Bietti, G. Ercolani and M. Salamone, Tetrahedron, 2002, 58, 5039 Search PubMed.
- (a) E red[TCB] −0.66 V vs. SCE; Eexc[1TCB] 4.10 eV, see Photoinduced Electron Transfer, Vol. 1 (ed. M. Chanon, L. Eberson, M. A. Fox, M. Chanon) Elsevier, Amsterdam, 1988, p. 409 Search PubMed; (b) E ox[cyclohexane] 3.17 V vs.SCE, calculated through the Miller relation from the measured IP, Z. B. Alfassi, R. E. Huie, J. P. Mittal, P. Neta and L. C. T. Shoute, J. Phys. Chem., 1993, 97, 9120 Search PubMed; (c) E ox[1-dioxane] 2.89 V vs.SCE, H. Ishii, Y. Hou and T. Fuchigami, Tetrahedron, 2000, 56, 8877 Search PubMed; (d) E ox[triethylamine] 1.07 V vs.SCE, T. Mizushima, S. Ikeda, S. Murata, K. Ishii and H. Hamaguchi, Chem. Lett., 2000, 1282 Search PubMed.
- (a) A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551 CrossRef CAS; (b) M. Mella, M. Freccero, E. Fasani and A. Albini, Chem. Soc. Rev., 1998, 27, 81 RSC; (c) P. R. Schreiner and A. A. Fokin, Chem. Rec., 2004, 3, 247 CrossRef CAS.
- (a) P. R. Schreiner, A. Wittkopp, P. A. Gunchenko, A. I. Yaroshinsky, S. A. Peleshanko and A. A. Fokin, Chem.–Eur. J., 2001, 7, 2739 CrossRef CAS; (b) A. A. Fokin, B. A. Tkachenko, T. A. Shubina, P. A. Gunchenko, D. V. Gusev, J. K. Vohs, G. H. Robinson, A. G. Yurchenko and P. R. Schreiner, Eur. J. Org. Chem., 2002, 3844 Search PubMed.
- M. Fagnoni, M. Mella and A. Albini, Tetrahedron, 1995, 51, 859 CrossRef CAS.
- (a) K. Toriyama, K. Nunome and M. Iwasaki, J. Am. Chem. Soc., 1987, 109, 4496 CrossRef CAS; (b) K. Toriyama, K. Nunome and M. Iwasaki, J. Phys. Chem., 1986, 90, 6836 CrossRef CAS; (c) P. A. Gunchenko, A. M. Makukhina, A. A. Novikovskii, A. G. Yurchenko, M. Serafin, P. R. Schreiner and A. A. Fokin, Theor. Exp. Chem., 2009, 45, 246 Search PubMed.
- G. Buntinx, R. Naskrecki and O. Poizat, J. Phys. Chem., 1996, 100, 19380 CrossRef CAS.
- (a) T. Shiba, in Electronic Absorption Spectra of Organic Radical Ions, Elsevier, Amsterdam, 1988 Search PubMed; (b) S. Nagara, in Excited States, Vol. 2, (Ed. E. C. Lim) Academic Press. 1995, 336 Search PubMed; (c) Both the cyclohexane and the 1,4-dioxane radical cations seem to absorb weakly and are not detected under these conditions, see I. A. Shkrob, M. C. Sauer and A. D. Trifunac, J. Phys. Chem., 1996, 100, 7237 Search PubMed; (d) H. Liu, Y. Hu, S. Yang, W. Guo, Q. Fu and L. Wang, J. Phys. Chem. A, 2006, 110, 4389 Search PubMed; (e) Likewise, the triethyamine radical cation shows a diffuse absorption over the visible that does not interfere with the strong spectrum of TCB˙−, see T. Shiba, Y. Nosaka and T. Kato, J. Phys. Chem., 1978, 82, 695 Search PubMed; (f) O. Poizat, G. Buntinx and L. Boilet, J. Phys. Chem. A, 2005, 109, 10813–10823 CrossRef CAS.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311 CrossRef CAS.
- S. A. Kovalenko, R. Schanz, H. Hennig and N. P. Ernsting, J. Chem. Phys., 2001, 115, 3256 CrossRef CAS.
- M. Fagnoni, M. Vanossi, M. Mella and A. Albini, Tetrahedron, 1996, 32, 1785 Search PubMed.
- M. Taguchia, Y. Matsumoto, M. Moriyama, H. Namba, Y. Aokia and H. Hiratsuka, Radiat. Phys. Chem., 2000, 58, 123 Search PubMed.
- (a) T. Lund, P. Christensen and R. Wilbrandt, Org. Biomol. Chem., 2003, 1, 1020 RSC; (b) S. U. Pedersen, T. Lund, K. Daasbjerg, M. Pop, I. Fussing and H. Lund, Acta Chem. Scand., 1998, 52, 657 CAS.
- (a) The enthalpy for the cleavage of a radical cation RH˙+ → R˙ + H+ was estimated from a thermochemical cycle, ΔH[RH˙+] = ΔH[RH] + F[Eox[H˙] − Eox[RH] and the entropy term was neglected; compare ref. 2d and 18b. For cyclohexane the calculated BDE of the radical cation [RH˙+ → R˙] was negative, −27.5 kcal mol−1; in triethylamine the BDE of the α C–H bond was reduced to 6 kcal mol−1, see ref. 15c.; (b) D. D. M. Wayner, D. J. McPhee and D. Griller, J. Am. Chem. Soc., 1988, 110, 132 CrossRef CAS; (c) D. D. M. Wayner, K. B. Clark, A. Rauk, D. Yu and D. A. Armstrong, J. Am. Chem. Soc., 1997, 119, 8925 CrossRef CAS.
- X. Zhang, S.-R. Yeh, S. Hong, M. Freccero, A. Albini, D. E. M. Falvey and S. Patrick, J. Am. Chem. Soc., 1994, 116, 4211 CrossRef CAS.
- F. D. Lewis, Acc. Chem. Res., 1986, 19, 401 CrossRef CAS.
- E ox[Et2NCH˙CH3] = −1.12 V vs.SCE, Eox[1[1,4-dioxolan-2-yl] = −0.08 V vs.SCE, see ref. 18b and D. D. M. Wayner and V. D. Parker, Acc. Chem. Res., 1993, 26, 287 Search PubMed.
- (a) For analogous ET processes, see e.g. M. Dossot, X. Allonas and P. Jacques, J. Photochem. Photobiol., A, 1999, 128, 47 Search PubMed; (b) C. Devadoss and R. W. Fessenden, J. Phys. Chem., 1991, 95, 7253 CrossRef CAS.
- H. Yokoi, S. Moriizumi, K. Ishiguro and Y. Sawaki, J. Photochem. Photobiol., A, 1999, 125, 39 CrossRef CAS.
- The rate constant of addition of an alkyl radical onto an electron-poor olefin is in the order of 106 M−1 s−1. See: H. Fischer and L. Radom, Angew. Chem., Int. Ed., 2001, 40, 1340 Search PubMed.
- (a) M. Freccero, M. Mella and A. Albini, Tetrahedron, 1994, 50, 2115 CrossRef CAS; (b) M. A. Kellet, D. G. Whitten, I. R. Gould and W. R. Bergmark, J. Am. Chem. Soc., 1991, 113, 358 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Absorption and fluorescence spectra of TCB, femtosecond absorption spectra of TCB under different conditions and kinetic analyses of obtained data. See DOI: 10.1039/c2ra01054b |
| This journal is © The Royal Society of Chemistry 2012 |