DOI:
10.1039/C2RA01107G
(Paper)
RSC Adv., 2012,
2, 3823-3827
Sol–gel synthesis of Au/N–TiO2 composite for photocatalytic reduction of Cr(VI)
Received
16th November 2011
, Accepted 30th January 2012
First published on 7th March 2012
Abstract
Au/N–TiO2 composites are successfully synthesized via a modified sol–gel method and their photocatalytic performance in reduction of Cr(VI) is investigated. Au/N–TiO2 composites exhibit an enhanced photocatalytic performance in the reduction of Cr(VI) with a maximum reduction rate of 90% under visible light irradiation as compared with pure TiO2 (34%) and N–TiO2 (80%) due to the increase of light absorption intensity and range as well as the reduction of electron–hole pair recombination in TiO2 with the incorporation of N and Au.
1. Introduction
TiO2 has attracted tremendous attention as a promising photocatalyst for widespread environmental applications owing to its intriguing optical and electric properties.1–9 However, its universal use is restricted to ultraviolet (UV) light due to its intrinsically wide band gap (3.2 eV in the anatase phase), and the quick recombination of photo-induced electron–hole pairs has significantly decreased the photocatalytic performance of TiO2. Recent progress has shown that the co-doping of TiO2 by non-metal elements,10–12 typically N or B, and metal nanomaterials with a work function lower than the conduction band of TiO2,13–18 such as Au, Ag or Fe, can solve these problems and enhance the photocatalytic activity of TiO2 in the UV or visible light region. In the photocatalysis process, metal nanomaterials can act as excellent electron-acceptor/transport materials to effectively facilitate the migration of photo-induced electrons and hinder the charge recombination in electron-transfer processes due to the electronic interaction between TiO2 and them, which enhances the photocatalytic performance,19,20 while non-metal elements can tune the band gap and extend the photo-response of TiO2 into the visible light region,21–25 which is regarded as an effective approach for the utilization of solar light. Wu et al.26 studied the synthesis of the Au/N–TiO2 composite and investigated its photocatalytic activity in the degradation of methyl orange. A two-step synthesis of N–TiO2 was carried out by the hydrolysis of titanium sulfate by ammonia followed by urea. Then, Au particles were deposited on the surface of N–TiO2 by using the deposition-precipitation method with urea. When Au was introduced into the N–TiO2 composite, the photocatalytic degradation rate of methyl orange increased from about 36% for N–TiO2 to 85.4% for Au/N–TiO2 (4 wt% Au) after reaction for 5 h under visible light irradiation. Parida et al.27 studied the synthesis of Au/S,N–TiO2 composite via a one-pot template-free homogeneous co-precipitation technique and borohydrate reduction method, and the composite achieved a higher catalytic activity in the oxidation of carbon monoxide compared to Au/TiO2, which was attributed to the presence of oxygen vacancies and the creation of new adsorption sites for the adsorption and activation of O2 molecules. Tian et al.13 carried out a simple wet-chemical method to fabricate the Au/N–TiO2 composite which exhibited a much higher visible-light photocatalytic activity in the degradation of 2,4-dichlorophenol compared with single N-doped or Au-loaded TiO2 due to the synergetic effects of N doping and Au loading. Iliev et al.28 reported that Au/N–TiO2 synthesized through a sol–gel and photo-reduction method improved the photocatalytic oxidation rate of oxalic acid, which was ascribed to the more efficient charge separation, the increase in the lifetime of the charge carriers and the enhancement of the efficiency of the interfacial charge transfer to adsorbed substrates. Despite the above progress to date, the exploration on Au/N–TiO2 composites, especially on their synthesis via the sol–gel method and application in photocatalytic reduction of Cr(VI), is not nearly enough. In this work, the one-step synthesis of Au/N–TiO2 composites is carried out via the hydrolysis of tetrabutyl titanate in HAuCl4 and urea solution to form a transparent sol. Au/N–TiO2 composites exhibit an enhanced photocatalytic performance in the reduction of Cr(VI) with a maximum reduction rate of 90% after reaction for 4 h under visible light irradiation as compared with pure TiO2 (34%) and N–TiO2 (80%).
2. Experimental
2.1 Synthesis of TiO2–N/Au composite
4 ml tetrabutyl titanate was first dissolved in 10 ml ethanol by stirring for 30 min at room temperature to obtain solution A. A certain amount of HAuCl4 and 429 mg urea were dissolved in 7 ml ethanol, and then 2 ml deionized water and 5 ml acetic acid were successively added to the solution with stirring for 30 min at room temperature to obtain solution B. Solution B was then added dropwise into solution A under vigorous stirring. Subsequently, the mixture solution was continuously stirred at 40 °C for the hydrolysis of tetrabutyl titanate until a transparent sol was formed. Finally, the sol was dried in air at 100 °C for 24 h, ground and heated at 500 °C for 1 h. The as-synthesized Au/N–TiO2 samples with 0, 0.1, 0.3, 0.5 wt% Au were named as TNA-0, TNA-1, TNA-2, and TNA-3, respectively. The pure TiO2 was also synthesized for comparison. For the electrochemical impedance spectra (EIS) testing, the as-synthesized composites with 5 wt% cellulose binder were homogenously mixed in terpineol to form a slurry. Then, the resultant slurries were coated on the FTO using a screen-printing approach. Finally, these prepared electrodes were sintered at 500 °C for 30 min.
2.2 Characterization
The surface morphology, structure and composition of the samples were characterized by field-emission scanning electron microscopy (FESEM, Hitachi S-4800), high-resolution transmission electron microscopy (HRTEM, JEOL-2010), X-ray diffraction spectroscopy (XRD, Holland Panalytical PRO PW3040/60) with Cu-Kα radiation (V = 30 kV, I = 25 mA), and energy dispersive X-ray spectroscopy (EDS, JEM-2100), respectively. The UV-vis absorption spectra were recorded using a Hitachi U-3900 UV-vis spectrophotometer. EIS measurement was carried out on an electrochemical workstation (AUTOLAB PGSTAT302N) under dark conditions using a three-electrode configuration with the as-prepared films as working electrode, a Pt foil as counter electrode and a standard calomel electrode as reference electrode. The electrolyte was 10 mg l−1 K2Cr2O7 aqueous solution. EIS were recorded in the frequency range of 0.1 Hz to 1 MHz, and the applied bias voltage and ac amplitude were set at open-circuit voltage and 10 mV AC between the counter electrode and the working electrode, respectively.
2.3 Photocatalytic experiments
The photocatalytic performance of the as-prepared samples was evaluated by photocatalytic reduction of Cr(VI) under visible light irradiation. The samples (1 g l−1) were dispersed in 60 ml Cr(VI) solutions (10 mg l−1) which were prepared by dissolving K2Cr2O7 into deionized water. The mixed suspensions were first magnetically stirred in the dark for 0.5 h to reach the adsorption–desorption equilibrium. Under ambient conditions and stirring, the mixed suspensions were exposed to visible light irradiation produced by a 400 W metal halogen lamp with cut off filter (λ > 400 nm). At certain time intervals, 2 ml of the mixed suspensions were extracted and centrifugated to remove the photocatalyst. The filtrates were analyzed by recording UV-vis spectra of Cr(VI) using a Hitachi U-3900 UV-vis spectrophotometer.
3. Results and discussion
Fig. 1(a)–(e) show the FESEM images of TiO2, TNA-0, TNA-1, TNA-2, and TNA-3, respectively. It is clearly seen that all the samples display spherical particles and that the average particle size decreases with the increase of Au content, showing that Au loading inhibits the growth of TiO2 crystallites.29 A decrease in particle size can lead to a larger surface area and lower volume recombination, which can facilitate the adsorption of more pollutants, and thus improve the photocatalytic performance.30 The existence of N-TiO2 in the composite is proved by the peaks of Ti, O and N in EDS spectra of TNA-3 (Fig. 1(f)). However, no diffraction peak of Au is observed, which may be due to the low amount of Au in the composite.
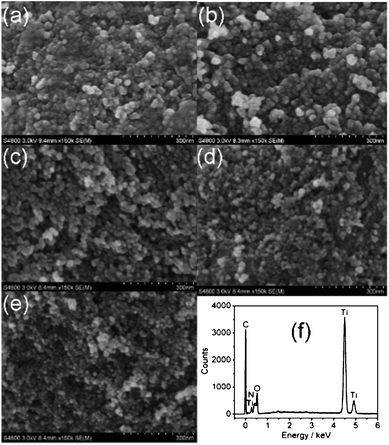 |
| | Fig. 1 Surface morphologies of (a) TiO2, (b) TNA-0, (c) TNA-1, (d) TNA-2, and (e) TNA-3 by FESEM measurement; (f) EDS spectrum of TNA-3. | |
Fig. 2(a) and (b) show the low-magnification and high-magnification HRTEM images of TNA-3. It can be observed that Au nanoparticles have attached onto the surface of TiO2. The large crystallite is identified as the TiO2 particle. The lattice spacing measured for this crystalline plane is 0.352 nm, corresponding to the (101) plane of anatase TiO2. Around the TiO2 crystallite edge, fine crystallites are observed. The crystallites connected to the TiO2 have lattice fringes of 0.236 nm which is ascribed to the (111) plane of Au.
 |
| | Fig. 2 (a) Low-magnification and (b) high-magnification HRTEM images of TNA-3. | |
The XRD patterns of TiO2, TNA-0, TNA-1, TNA-2, and TNA-3 are shown in Fig. 3. It can be observed that all the samples exhibit the diffraction peaks at 25.3°, 37.8°, 48.0°, 53.9°, 55.1°, 62.7°, 68.8°, 70.3°, and 75.0°, indexed to (101), (004), (200), (105), (211), (204), (116), (220), and (215) crystal planes of anatase TiO2 (JCPDS 21-1272), which demonstrates that the co-doping of N and Au does not result in the development of new crystal orientations of TiO2. Compared with pure TiO2 and TNA-0, new peaks at 44.4°, 64.6°, and 77.6°, corresponding to the (200), (220) and (311) planes of polycrystalline Au (JCPDS 76-1802), appear in XRD pattern of TNA-2 and TNA-3, which further confirm the existence of Au in the composite. It should be noticed that the absence of the Au peak for TNA-1 should be ascribed to the low amount of Au being below the detection limit.
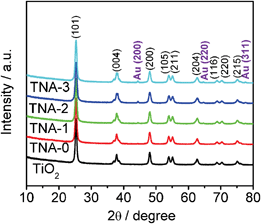 |
| | Fig. 3 XRD patterns of TiO2, TNA-0, TNA-1, TNA-2, and TNA-3. | |
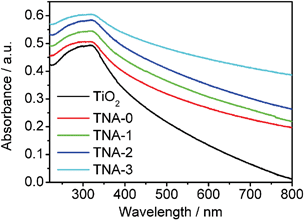
The UV-vis absorption spectra of TiO2, TNA-0, TNA-1, TNA-2, and TNA-3 are shown in Fig. 4. It is observed that TiO2 presents its characteristic absorption peak at 330 nm and the absorbance of Au/N–TiO2 composites increases even in visible light region with the increase of Au content, which is similar to other reported results.13,26,31 Such an enhancement in absorbance can increase the number of photo-generated electrons and holes to participate in the photocatalytic reaction and enhance the photocatalytic performance.32 In addition, the absorption edge of the Au/N–TiO2 composite exhibits a red shift toward the visible light region compared with TiO2, which is attributed to band gap narrowing by the N doping.33 Similar results are also observed in other TiO2-based composite materials.34,35 Therefore, the incorporation of N and Au in TiO2 can increase the visible light absorption intensity and range, which is beneficial to the photocatalytic performance.
The photocatalytic reduction of Cr(VI) under visible light irradiation was used to evaluate the photocatalytic performance of TiO2, TNA-0, TNA-1, TNA-2, and TNA-3, as shown in Fig. 5. The normalized temporal concentration changes (C/C0) of Cr(VI) during the photocatalytic process are proportional to the normalized maximum absorbance (A/A0), which is derived from the changes in the Cr(VI) absorption profile at a given time interval. It can be observed that TNA-0 composite exhibits much higher photocatalytic reduction rate (80%) than pure TiO2 (34%) because the doping of N extends the absorption of TiO2 into the visible light range,33,36 and inhibits the recombination of the charge carriers.37 When Au is introduced into the composite, the removal rate is increased to 87% for TNA-1 and reaches a maximum value of 90% for TNA-2. It indicates that the incorporation of N and Au into TiO2 play an important role in the photocatalytic performance. It is known that during photocatalysis, the adsorption of pollutants, the light absorption and the charge transportation and separation are crucial factors.38–40 The enhancement of the photocatalytic performance should be ascribed to the increase of the adsorption of pollutants, the increase of the visible light absorption intensity and range as well as the reduction of electron–hole pair recombination in TiO2 with the incorporation of N and Au. As shown in Fig. 6, the typical EIS of the pure TiO2, TNA-0, and TNA-2 electrodes are presented as Nyquist plots. It is observed that the semicircle in the plot becomes shorter with the incorporation of N and Au into TiO2, indicating a favored electron transport and the reduction of electron recombination which is related to the stepwise structure of energy levels constructed in the Au/N-TiO2 composite, as shown in Fig. 7. The conduction band and valence band of TiO2 are −4.2 eV and −7.4 eV (vs. vacuum).41 The work function of Au is around −5.1 eV.42 Such energy levels are beneficial for photo-induced electrons to transfer from the TiO2 conduction band to Au, which could efficiently separate the photo-induced electrons and hinder the charge recombination in electron-transfer processes,26,43 and thus enhance the photocatalytic performance. However, when the Au content is further increased above its optimum value, the photocatalytic performance deteriorates. This is ascribed to the following reasons: (i) Au can absorb some visible light19,26,31,44 and thus there exists a light harvesting competition between N–TiO2 and Au with the increase of Au amount, which leads to the decrease of the photocatalytic performance; (ii) the excessive Au can act as a kind of recombination center instead of providing an electron pathway and promote the recombination of electron–hole pairs in Au.13,19,45
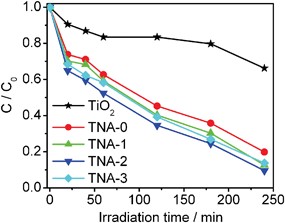 |
| | Fig. 5 Photocatalytic reduction of Cr(VI) by TiO2, TNA-0, TNA-1, TNA-2, and TNA-3 under visible light irradiation. The concentrations of Cr(VI) and photocatalyst are 10 mg l−1 and 1 g l−1, respectively. | |
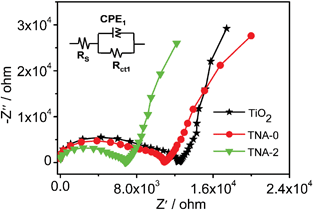 |
| | Fig. 6 Nyquist plots of the pure TiO2, TNA-0, and TNA-2 electrodes. Inset is the corresponding equivalent circuit model. | |
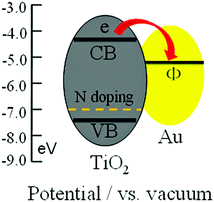 |
| | Fig. 7 Schematic diagram of energy levels of N–TiO2 and Au. CB, VB and Φ are conduction band, valence band and work function, respectively. | |
4. Conclusions
Au/N–TiO2 composites are successfully synthesized via a simple sol–gel method and their photocatalytic performance is investigated. The experimental results indicate that (i) Au/N–TiO2 composites exhibit a better photocatalytic performance than pure TiO2 and N–TiO2; (ii) the photocatalytic performance of the Au/N–TiO2 composite is dependent on the proportion of Au in the composite, and the composite with 0.3 wt% Au achieves a highest reduction rate of 90%; (iii) the enhanced photocatalytic performance is mainly ascribed to the increase of visible light absorption intensity and range as well as the reduction of photoelectron–hole pair recombination in TiO2 with the incorporation of N and Au.
Acknowledgements
Financial support from Special Project for Nanotechnology of Shanghai (No. 1052nm02700) and ECNU Reward for Excellent Doctors in Academics (No. XRZZ2011017) is gratefully acknowledged.
References
- N. Murakami, Y. Fujisawa, T. Tsubota and T. Ohno, Appl. Catal. B: Environ., 2009, 92, 56 CrossRef CAS.
- T. Tsubota, A. Ono, N. Murakami and T. Ohno, Appl. Catal. B: Environ., 2009, 91, 533 CrossRef CAS.
- Z. Xiong, H. Dou, J. Pan, J. Ma, C. Xu and X. S. Zhao, CrystEngComm., 2010, 12, 3455 RSC.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, Nanoscale, 2011, 3, 3670 RSC.
- S. B. Rawal, A. K. Chakraborty, Y. J. Kim, H. J. Kim and W. I. Lee, RSC Adv., 2012, 2, 622 RSC.
- M. A. Lazar and W. A. Daoud, RSC Adv., 2012, 2, 447 RSC.
- Y. L. Liao and W. X. Que, J. Alloys Compd., 2010, 505, 243 CrossRef CAS.
- Y. L. Liao, W. X. Que, Z. H. Tang, W. J. Wang and W. H. Zhao, J. Alloys Compd., 2011, 509, 1054 CrossRef CAS.
- F. Y. Shen, W. X. Que, Y. L. Liao and X. T. Yin, Ind. Eng. Chem. Res., 2011, 50, 9131 CrossRef CAS.
- X. S. Zhou, F. Peng, H. J. Wang, H. Yu and J. Yang, Electrochem. Commun., 2011, 13, 121 CrossRef CAS.
- M. Y. Xing, J. L. Zhang and F. Chen, J. Phys. Chem. C, 2009, 113, 12848 CAS.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, Phys. Chem. Chem. Phys., 2011, 13, 4853 RSC.
- B. Z. Tian, C. Z. Li, F. Gu and H. B. Jiang, Catal. Commun., 2009, 10, 925 CrossRef CAS.
- S. S. Zhang, F. Peng, H. J. Wang, H. Yu, S. Q. Zhang, J. Yang and H. J. Zhao, Catal. Commun., 2011, 12, 689 CrossRef CAS.
- M. Y. Xing, Y. M. Wu, J. L. Zhang and F. Chen, Nanoscale, 2010, 2, 1233 RSC.
- V. Iliev, D. Tomova, S. Rakovsky, A. Eliyas and G. L. Puma, J. Mol. Catal. A: Chem., 2010, 327, 51 CrossRef CAS.
- E. Kowalska, R. Abe and B. Ohtani, Chem. Commun., 2009, 241 RSC.
- Z. Xiong, J. Ma, W. J. Ng, T. D. Waite and X. S. Zhao, Water Res., 2011, 45, 2095 CrossRef CAS.
- M. V. Dozzi, L. Prati, P. Canton and E. Selli, Phys. Chem. Chem. Phys., 2009, 11, 7171 RSC.
- X. D. Wang and R. A. Caruso, J. Mater. Chem., 2011, 21, 20 RSC.
- J. Zhong, F. Chen and J. L. Zhang, J. Phys. Chem. C, 2009, 114, 933 Search PubMed.
- W. Zhang, L. D. Zou and L. Z. Wang, Chem. Eng. J., 2011, 168, 485 CrossRef CAS.
- A. G. Kontos, M. Pelaez, V. Likodimos, N. Vaenas, D. D. Dionysiou and P. Falaras, Photochem. Photobiol. Sci., 2011, 10, 350 CAS.
- V. M. Menendez-Flores, D. W. Bahnemann and T. Ohno, Appl. Catal. B: Environ., 2011, 103, 99 CrossRef CAS.
- M. Bellardita, M. Addamo, A. Di Paola, L. Palmisano and A. M. Venezia, Phys. Chem. Chem. Phys., 2009, 11, 4084 RSC.
- Y. M. Wu, H. B. Liu, J. L. Zhang and F. Chen, J. Phys. Chem. C, 2009, 113, 14689 CAS.
- K. M. Parida, N. Sahu, A. K. Tripathi and V. S. Kamble, Environ. Sci. Technol., 2010, 44, 4155 CrossRef CAS.
- V. Iliev, D. Tomova and S. Rakovsky, Desalination, 2010, 260, 101 CrossRef CAS.
- P. Wongwisate, S. Chavadej, E. Gulari, T. Sreethawong and P. Rangsunvigit, Desalination, 2011, 272, 154 CrossRef CAS.
- Z. B. Zhang, C. C. Wang, R. Zakaria and J. Y. Ying, J. Phys. Chem. B, 1998, 102, 10871 CrossRef CAS.
- X. F. Li, T. X. Fan, H. Zhou, B. Zhu, J. Ding and D. Zhang, Micropor. Mesopor. Mater., 2008, 116, 478 CrossRef CAS.
- M. Hamadanian, A. Reisi-Vanani and A. Majedi, Appl. Surf. Sci., 2010, 256, 1837 CrossRef CAS.
- M. Y. Xing, J. L. Zhang and F. Chen, Appl. Catal. B: Environ., 2009, 89, 563 CrossRef CAS.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715 CAS.
- B. Naik, K. M. Parida and C. S. Gopinath, J. Phys. Chem. C, 2010, 114, 19473 CAS.
- Y. Wang, C. X. Feng, M. Zhang, J. J. Yang and Z. J. Zhang, Appl. Catal. B: Environ., 2011, 104, 268 CrossRef CAS.
- J. X. Yuan, E. J. Wang, Y. M. Chen, W. S. Yang, J. H. Yao and Y. A. Cao, Appl. Surf. Sci., 2011, 16, 7335 CrossRef.
- H. Zhang, X. J. Lv, Y. M. Li, Y. Wang and J. H. Li, ACS Nano, 2010, 4, 380 CrossRef CAS.
- X. J. Liu, L. K. Pan, T. Lv, T. Lu, G. Zhu, Z. Sun and C. Q. Sun, Catal. Sci. Technol., 2011, 1, 1189 CAS.
- X. J. Liu, L. K. Pan, T. Lv, G. Zhu, T. Lu, Z. Sun and C. Q. Sun, RSC Adv., 2011, 1, 1245 RSC.
- M. Gratzel, Nature, 2001, 414, 338 CrossRef CAS.
- B. Kouskoussa, M. Morsli, K. Benchouk, G. Louarn, L. Cattin, A. Khelil and J. C. Bernede, Phys. Status Solidi A, 2009, 206, 311 CrossRef CAS.
- G. Zhu, F. F. Su, T. Lv, L. K. Pan and Z. Sun, Nanoscale Res. Lett., 2010, 5, 1749 CrossRef CAS.
- Z. K. Zheng, B. B. Huang, X. Y. Qin, X. Y. Zhang, Y. Dai and M. H. Whangbo, J. Mater. Chem., 2011, 21, 9079 RSC.
- J. G. Yu, L. Yue, S. W. Liu, B. B. Huang and X. Y. Zhang, J. Colloid Interface Sci., 2009, 334, 58 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.