Ligand density and clustering effects on endocytosis of folate modified nanoparticles†
Emilia
Moradi
a,
Driton
Vllasaliu
a,
Martin
Garnett
a,
Franco
Falcone
b and
Snow
Stolnik
*a
aDivision of Drug Delivery and Tissue Engineering, School of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: snjezana.stolnik@nottingham.ac.uk; Fax: (+) 44 115 9515102; Tel: (+) 44 115 8466074
bMolecular and Cellular Science Division, School of Pharmacy, University of Nottingham, NG7 2RD, UK
First published on 17th February 2012
Abstract
This study investigates the effects of surface ligand distribution pattern (ligand clustering and density) on the internalisation of nanoparticles by a bronchial epithelial in vitro model (Calu-3 cells cultured as polarised layers). Control of ligand clustering and its surface density was achieved through the use of ovalbumin as an intermediate species to anchor the ligand to the nanoparticle surface. The model particulate system consisted of polystyrene nanoparticles surface-decorated via the adsorption of ovalbumin with conjugated folate ligand. The density of the displayed ligand was manipulated by controlling the conjugation level of folate to ovalbumin, while ligand clustering was achieved by co-adsorption of varying mixtures of folate-ovalbumin conjugate (at different ligand density levels) and unconjugated ovalbumin. Increasing overall ligand density on the nanoparticle surface resulted in increased internalisation of modified nanoparticles by the cells, up to a saturation level. Surface ligand density also affected the cellular uptake pathway; from predominantly clathrin to predominantly caveolae-mediated as the ligand density is increased. We further demonstrate that surface clustering of the folate ligand enhances cellular internalisation of nanoparticles, relative to its dispersed surface distribution. Our work suggests a simple way to prepare a model system where surface manipulations of ligand density and its distribution are possible and which can be used to study nanoparticle–cell interaction processes.
Introduction
To achieve intracellular delivery of biologicals (proteins, oligonucleotides or siRNA), the crucial step is to design a delivery system that both protects the incorporated biologic and is efficiently internalised by the cells of interest. The approaches used to formulate delivery systems and achieve cellular targeting and internalisation are typically based on sub-micron sized particulate ‘carriers’, such as liposomes or polymeric nanoparticles which are decorated on their surfaces with targeting ligands, based on either antibodies, polypeptides, fusogenic proteins or hormones. The choice of ligand depends on the type(s) of cells/tissues to be targeted. After attachment, the targeting ligand must be exposed and accessible at the carrier surface to ensure effective interaction with the receptor at the targeted cell. For intracellular delivery, it is also important that the ligand–receptor binding interaction leads to internalisation of the carrier, followed by an appropriate intracellular transport route, which will eventually deliver a therapeutic biologic to the target intracellular site.The density of a ligand on the surface of a particulate carrier can have a profound effect on ligand–receptor binding events and the biological processes that follow this binding. Studies on this phenomenon have recently taken momentum with a general aim to enhance the prospects of targeting systems by improving their binding and cellular uptake by targeted cells. Surface density of different ligands has been demonstrated to play an important role in the cellular uptake of, for instance, PEG-PLA nanoparticles, surface modified with wheat germ agglutinin1 or PLGA nanoparticles modified with a cyclic peptide that specifically binds to ICAM-1 (intercellular cell adhesion molecule-1),2 composite polymeric micelles surface decorated with RNA A10 aptamer which specifically binds to the prostate-specific membrane receptor,3 liposomes surface modified with fibronectin-mimetic peptide4 and caprolactone-polyethylene glycol micellar nanoparticles surface modified with bovine serum albumin as a model ligand.5 The influence of ligand surface density on cell uptake has also been demonstrated in our recent work with composite polymeric micelles surface decorated with the folate ligand.6
Typically, the surface density of the ligand is controlled by adjusting the composition of the mixture containing ligand-modified and unmodified polymeric molecules that either self-assemble into a micellar-type nanocarrier3 or adsorb at the surface of nanoparticles.2 However, such approaches to achieve surface ligand presentation typically do not allow control over its spatial distribution and consequently, the effect of ligand surface patterning on the binding and cellular uptake remains largely unstudied.7 Scarce publications on the effect of ligand clustering, using branched materials that afford multi-ligand attachment per polymeric molecule, indicate potentially important implications for cell targeting and development of more effective targeting systems.8
In the present work we describe a model nanoparticle system which enables manipulation of the surface density as well as surface clustering of a ligand. This was achieved by controlling the level of ligand (folic acid) conjugation to an ‘anchor’ species (ovalbumin) which possesses multi-ligand attachment capacity. Subsequent adsorption of such ligand-anchor conjugate with different conjugation levels onto nanoparticle surface, alone or in the mixture with unconjugated species, allows control over surface density and clustering. The system provided a mean to test, not only the impact of surface ligand density, but also of ligand spatial distribution (clustering) at the carrier surface on its cellular internalisation behaviour.
Results and discussion
Nanoparticle preparation and characterization
Initially, folate ligand (FA) was conjugated to ovalbumin (OVA) as an ‘anchoring’ species that serves to achieve multi-ligand attachment, as well as to anchor the ligand to the surface of a polymeric carrier, in this case polystyrene latex, via adsorption. FA-OVA conjugates with varying molar substitution ratios (MSR) of 1.5 ± 0.2, 3.3 ± 2.1, 5.5 ± 0.2, 7.8 ± 1.6, 9.0 ± 0.8 and 13.2 ± 1.0 (mean ± S.D.; n = 6) mol FA per mol OVA were produced and adsorbed onto 30 nm polystyrene latex (as presented schematically in Fig. 1). Ovalbumin, rather than human serum albumin was used in the present work as a multi-ligand anchor for folate, as the latter has been reported to have specific uptake pathways in epithelial cells via the gp60 receptor,9 which would likely influence the cell uptake behaviour of nanoparticles. Ovalbumin is structurally significantly different from serum albumin and is not recognised by the same receptor mediated pathway.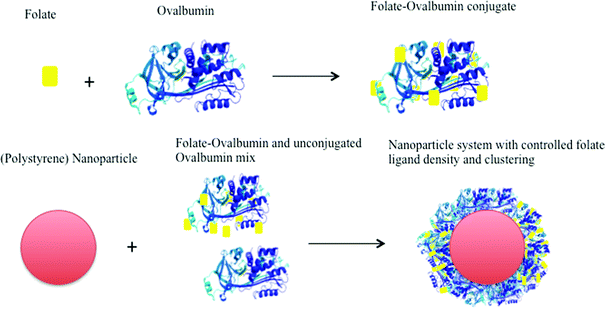 | ||
| Fig. 1 Preparation of folate-decorated polystyrene nanoparticles. Each ovalbumin ‘anchor’ molecule allows covalent linkage of multiple ligands. Co-adsorption of ligand-conjugated (at different molar substitution ratio) and unconjugated anchor molecules achieves ligand clustering on nanoparticle surface. | ||
Particle size determination of a series of FA-OVA decorated nanoparticles by dynamic light scattering is shown in Fig. 2. The systems in the series comprised of nanoparticles with surface adsorbed FA-OVA conjugates produced at different molar substitution ratios of folate to ovalbumin (Fig. 2C–F). For the purpose of comparison, the size of unmodified nanoparticles (i.e. prior to surface adsorption of FA-OVA conjugates) and following adsorption of unconjugated OVA are also shown (Fig. 2A and B). The particle distribution graphs show that there is no notable aggregation of the nanoparticles following surface modification. The mean hydrodynamic particle radius and distribution for all nanoparticle systems, surface decorated with unconjugated OVA (Fig. 2B) and those decorated with FA-conjugated OVA with varying molar substitution ratios (Fig. 2C–2F), ranged between 62–66 nm, indicating no significant differences in the particle size between modified nanoparticles that could potentially affect their cellular internalisation behaviour. Unmodified nanoparticles displayed a mean hydrodynamic particle radius of 17.9 nm (Fig. 2A), indicating the formation of an adsorbed layer in the order of 44–48 nm, considerably larger than what would be expected from adsorption of a monolayer of ovalbumin (molecular dimensions of 7 × 4.5 × 5 nm).10 Such behaviour would be in agreement with previous studies on ovalbumin adsorption onto a latex surface in which OVA adsorption occurs in excess of the quantity required to produce maximal coverage of the surface and where multilayer adsorption behaviour is indicated.11 In a similar way to hydrophobic sorption of albumin to polystyrene latex,12,13 the adsorption of OVA on the surface of polystyrene nanoparticles is expected to be irreversible under the conditions employed, permitting OVA (and hence ligand) association with the nanoparticles during the time course of the cell uptake experiments.
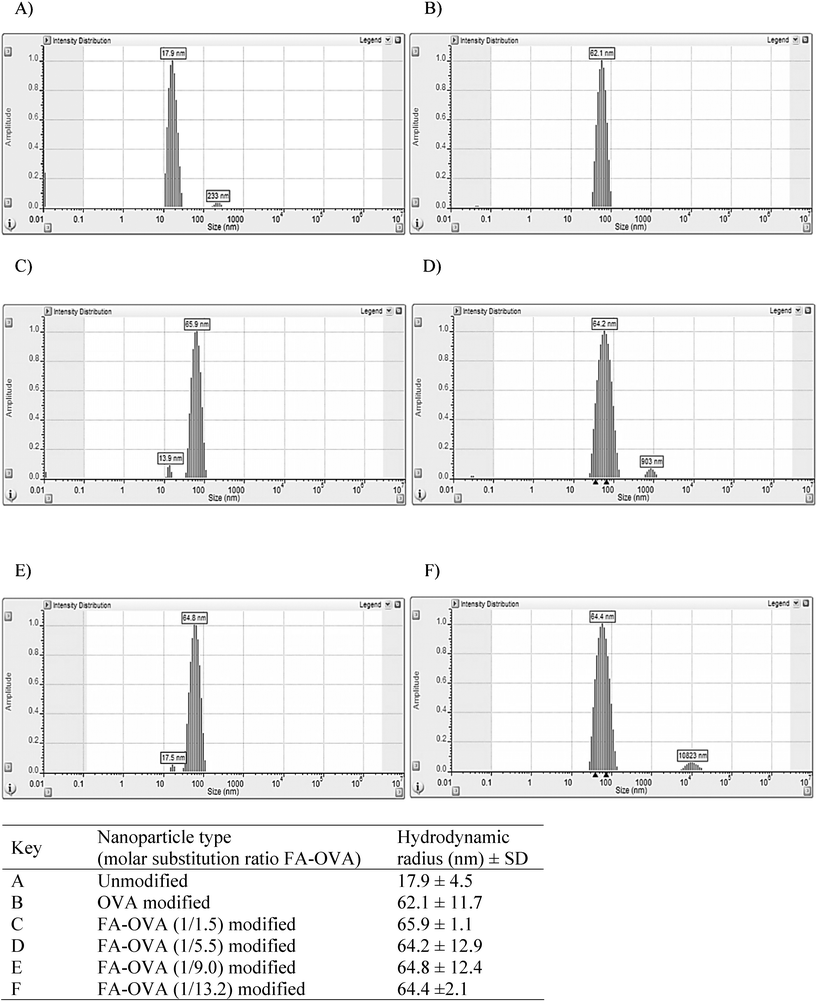 | ||
| Fig. 2 Particle size (hydrodynamic radius) and distribution of unmodified (A), and nanoparticles modified with either unconjugated ovalbumin (B) or folate-conjugated ovalbumin (FA-OVA) prepared at different molar substitution ratios (C, D, E and F), as determined by dynamic light scattering. The results in the table represent a mean ± SD of ten measurements performed in Hank's Balanced Salt Solution. | ||
In the present study the amount of conjugated or unconjugated OVA used per unit surface area of polystyrene nanoparticles (1.7 mg m−2) falls within the range of plateau adsorption values reported previously (e.g. approximately 1.5 mg m−214 and 1.2 mg m−2).15 The OVA molecules immobilised within the nanoparticle surface multilayer were assumed to adopt different orientations, including those that permit binding of conjugated folate ligand to the receptor and initiate nanoparticle cellular internalisation. By removing excess un-adsorbed ovalbumin (by centrifugation), we ensured that the possibility of free folate-OVA conjugates in the system competing for the folate receptors with the ligand-bearing nanoparticles was minimised.
Immunostaining for the folate receptor
Confocal micrographs of Calu-3 cell layers cultured on permeable supports, used as a model of the airway epithelium, and immunostained for the folate receptor (FOLR1) (Fig. 3) demonstrate the presence of positive signal for folate receptor (red fluorescence arising from TRITC-labelled secondary antibody), hence indicating expression of the folate receptor. The micrograph showing vertical axis (‘z-axis’) of the cell layer (located on the right side and bottom of the image) indicates that the receptor is distributed across the vertical plane of the cell layer. In the control cell layer treated with the secondary antibodies only, no fluorescence was observed (Fig. 3B), indicating that the fluorescence signal in the cell layer in Fig. 3A is not present due to non-specific secondary antibody binding. Previous publications have reported that low expression of folate ligands in humans is restricted to certain normal tissues, including lung, kidney, intestine, heart, liver, spleen, muscle and brain,16 whilst a significantly higher expression level is seen in folate positive malignant tissues.16,17 The present study further demonstrates the expression of a folate receptor in Calu-3 epithelial cells layers.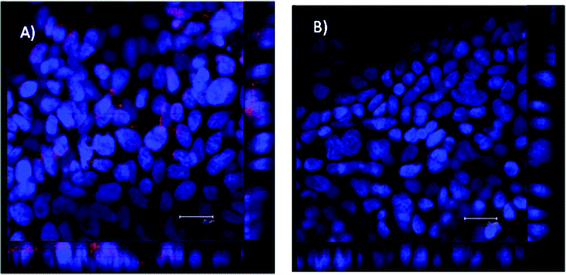 | ||
| Fig. 3 Confocal micrographs showing immunostaining for folate receptor (FOLR1) in filter-cultured Calu-3 cell layers. Cells were incubated with mouse, anti-human FOLR1 primary antibody and goat, anti-mouse TRITC (red)-labelled secondary antibodies (A). Calu-3 cell layer incubated with only goat, anti-mouse TRITC-labelled secondary antibody (control) (B). The image of the vertical (‘z-axis’) is shown on the bottom and the right side of the image. Scale bar = 20 μm. | ||
Effect of ligand density on cellular internalisation of nanoparticles
To study the effect of folate ligand density on cellular internalisation of nanoparticles, these were surface modified by adsorption of FA-OVA conjugates produced at different molar substitution ratios, ranging from 1.5 to 13.2 mol of folate per mol of OVA. Data in Fig. 4 clearly demonstrate that cell uptake of modified nanoparticles is affected by the surface density of the ligand (i.e. molar substitution ratio), whereby the uptake increases with an increase in folate ligand density and apparently reaches a plateau at high ligand densities. In comparison, control nanoparticles surface-modified with unconjugated OVA (therefore lacking in folate ligand) showed a significantly lower cell internalisation (p = 0.001). The trend seen in Fig. 4 is in close agreement with our previous study where a range of folate decorated micellar-type nanoparticles was produced by progressively increasing the content of folate functionalized copolymer relative to the ‘non-functionalized’ core polymer.6 A similar trend of increased folate ligand density resulting in increased internalisation by KB cells until an apparent plateau is reached was also reported for solid lipid nanoparticles with folate ligand conjugated to a lipid component incorporated into the formulation.18 A similar phenomenon was also reported for cell association of nanoparticles, with a plateau reached at higher ligand densities for nanosystems surface-modified with other ligands, for instance bacterial invasin (MBP-Inv479) attached at varying surface concentrations to 155–200 nm nanoparticles,19 or for RNA aptamer at the surface of micellar-type PLGA-PEG nanoparticles.3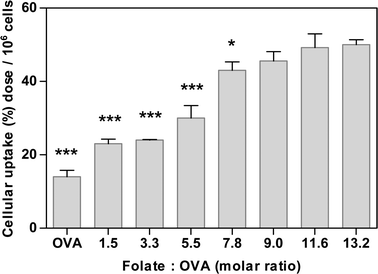 | ||
| Fig. 4 Effect of ligand density on cellular internalisation of nanoparticles in Calu-3 layers. Nanoparticles were surface modified with folate-ovalbumin (FA-OVA) conjugates produced at increasing FA to OVA molar substitution ratios (1.5–13.2). ‘OVA’ represents nanoparticles with unconjugated OVA (control). Uptake expressed as % of dose applied apically (containing 1.69 × 1013 nanoparticles) per one cell layer (average 1 × 106 cells). One-way analysis of variance (ANOVA) with Bonferroni post-hoc test was used for statistical analysis. Error bar represents mean ± standard deviation (n = 3). *, ** and *** denote p < 0.05, p < 0.01 and p < 0.001, respectively, relative to cell uptake of nanoparticles with FA-OVA at the highest molar substitution ratio of 13.2. | ||
The observed plateau in cell internalisation at higher ligand densities could arise from saturation of folate receptors at the cell surface,20 whereby increasing ligand density would increase the number of potential receptor interactions until a point at which the maximum number of interactions occurs and the rate of particle internalisation approaches the maximum rate allowed by the process, as previously suggested.19
Effect of surface ligand density on cell internalisation pathway (endocytosis)
To investigate the potential effect that surface density of ligand could have on the pathway(s) involved in cellular internalisation (endocytosis) of folate-decorated nanoparticles, clathrin and caveolae-mediated pathways were investigated as potential internalisation routes suggested for the folate ligand (Fig. 5).21–23 Sterol-binding agent filipin was applied to the Calu-3 cells to disrupt caveolae-mediated endocytosis,24 and cationic amphiphilic agent chlorpromazine to inhibit the clathrin-dependent pathway.25,26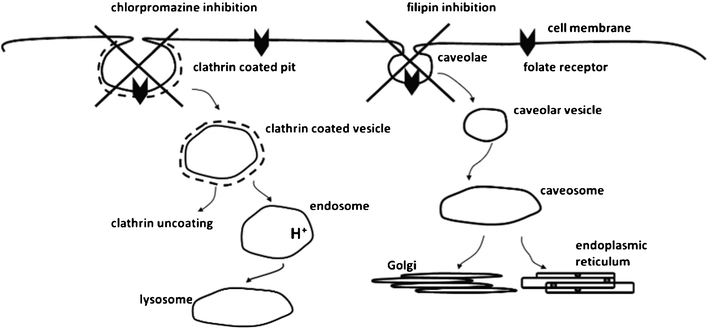 | ||
| Fig. 5 Cartoon representation of the possible cellular internalisation pathways suggested for folate ligand and their inhibition. Diagram is not to scale in representing the proportions of the different objects. | ||
In clathrin-mediated endocytosis, clathrin is first assembled at the intracellular surface of the plasma membrane to form a clathrin coated pit. The deeply invaginated coated pits pinch off from the plasma membrane to form a clathrin coated vesicle which loses its clathrin coat and progress to endosomes and subsequently towards lysosomes where the internalised material is degraded. For caveolae-mediated endocytosis, the caveolae (flask-shaped vesicles at the plasma membrane) invaginate and bud into the cytoplasm. The internalised caveolar vesicles fuse to form caveosomes. The internalised material is sorted to Golgi complex or endoplasmic reticulum.
Fig. 6 shows the impact of inhibitors of caveolae- or clathrin-mediated cell uptake pathways on internalisation of nanoparticles surface decorated with FA-OVA of varying substitution ratios. Considering the control system comprising of nanoparticles modified with unconjugated OVA, the cellular internalisation was notably reduced (p < 0.001) with chlorpromazine treatment (i.e. inhibition of clathrin pathway), whilst no significant reduction compared to the untreated cells was seen with filipin treatment (caveolae inhibition), clearly indicating that the cell uptake of folate-free nanoparticles is largely dependent on the clathrin-mediated pathway.
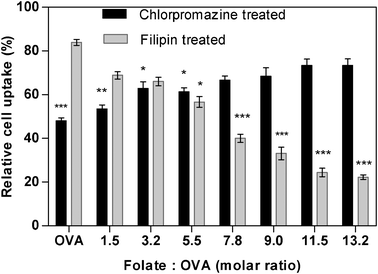 | ||
| Fig. 6 Cellular internalisation of folate-modified nanoparticles in the presence of endocytosis inhibitors for clathrin (chlorpromazine) or caveolae (filipin)-mediated pathways. Nanoparticles produced at increasing folate to ovalbumin molar substitution ratios (1.5–13.2). ‘OVA’ represent unconjugated ovalbumin modified nanoparticles. Uptake expressed as % relative to uptake of untreated cell layer. One-way analysis of variance (ANOVA) with Bonferroni post-hoc test was used for statistical analysis. Error bar represents mean ± standard deviation (n = 3). *p < 0.05, ** p < 0.01 and *** p < 0.001 relative to the respective uptake for cells untreated with the inhibitors. | ||
For FA-OVA modified nanoparticles, clathrin inhibition by chlorpromazine significantly (p < 0.01) influenced cell internalisation at the lowest surface density of the folate ligand, indicating that clathrin-mediated pathway is still dominant in the internalisation of this system. On the other hand, chlorpromazine-induced clathrin inhibition showed a moderate effect on systems with intermediate ligand densities (molar substitution ratios of 3.2 and 5.5; p < 0.05) and displayed no significant effect on the cellular uptake of nanoparticles at higher folate ligand densities (molar substitution ratios >7.8). A mirror trend of statistically significant decrease in cellular internalisation of nanoparticles at higher folate ligand densities (molar substitution ratios >7.8; p < 0.001) was apparent when caveolin-mediated endocytosis was inhibited by filipin. The suppression in cell uptake was modest at a lower ligand density (molar substitution ratio of 5.5) and insignificant at lowest two densities (molar substitution ratios 3.2 and 1.5). These findings indicate dominant involvement of caveolae-mediated internalisation of folate decorated nanoparticles and contribute to the body of literature where some studies point at caveolae21–23,27,28 and others at clathrin-mediated pathways for folate receptor uptake.29,30 The data further indicate a role that ligand density plays in determining internalisation pathway of folate-decorated nanoparticles.
Effect of ligand clustering on cellular internalisation
Table 1 summarises the data depicting the effect of ligand clustering on cell uptake of nanoparticles. A series of nanoparticle systems with different surface distribution of folate ligand were created by adsorption of different compositions of FA-conjugated OVA, with varying molar substitution ratios (1.5 to 13.2), in combination with unconjugated OVA. Variations in ligand density are largely determined by the folate molar substitution ratio (rows in Table 1), while clustering is largely determined by the composition of FA-OVA to unconjugated OVA mixtures (columns in Table 1).| Molar substitution ratio Folate : OVA | ||||||||
|---|---|---|---|---|---|---|---|---|
| Composition (%) of FA-OVA to OVA | 13.2 | 11.6 | 9.0 | 7.8 | 5.5 | 3.3 | 1.5 | |
| Amount of folate ligand used to modify nanoparticles (n mol) (in italics) | ||||||||
| Cellular internalisation (%) of applied dose (in bold) | ||||||||
| 100% | 26.7 | 23.3 | 18.2 | 15.8 | 11.1 | 6.5 | 2.9 | |
| 60.0 | 58.2 | ⁁153.4 | 49.0 | *242.3 | *129.4 | 24.1 | ||
| 75% | 19.9 | 17.4 | 13.6 | 11.8 | 8.3 | 4.9 | 2.1 | |
| 57.2 | *153.7 | *545.6 | *244.6 | *637.7 | *725.8 | *1013.2 | ||
| 50% | 13.4 | 11.6 | 9.1 | 7.9 | 5.5 | 3.2 | 1.4 | |
| *550.9 | *250.1 | *648.4 | *641.6 | *330.7 | *1021.5 | ⁁3*911.2 | ||
| 25% | 6.7 | 5.8 | 4.6 | 3.9 | 2.8 | 1.6 | 0.7 | |
| *138.2 | *334.6 | *730.4 | *1026.5 | *417.6 | *8⁁311.8 | 10.8 | ||
| 10% | 2.6 | 2.3 | 1.8 | 1.5 | 1.1 | 0.6 | 0.2 | |
| *4⁁224.2 | ⁁2*1021.1 | *818.8 | *918.2 | 15.3 | 12.4 | 10.0 | ||
At first, the table demonstrates dramatic differences in the level of nanoparticle internalisation by cells, extending between 60% of the applied dose for the highest surface content of folate ligand (referring to high molar substitution ratio of 13.2 at 100% FA-OVA) and 10% of the dose for the system with low folate content (referring to 1.5 molar substitution ratio and 10% FA-OVA mixture).
Combining this with the data in Fig. 6, showing that the pathway of nanoparticle uptake changes with the surface density of the ligand, the high ligand coverage systems will be taken into the cells at high levels (% of applied dose) and primarily by a caveolae-mediated pathway, whilst those with the low surface ligand coverage will be internalised in a considerably less efficient manner and primarily by a clathrin-mediated pathway. The matrix hence demonstrates how dramatically different the outcome, and potentially the drug delivery effect, could be regardless of the fact that the same ligand was employed for nanoparticle surface modification. In an in vivo situation further considerations would need to include effects that the opsonisation process would have on the nanoparticles’ surface and what would ultimately be presented to a cell to ‘see’ in such an environment.31,32
Comparing the systems where folate conjugates with the same molar substitution ratio in a mixture with unconjugated OVA were used to modify nanoparticles (along columns in the Table 1), the data clearly indicate that decreasing the overall surface density of the folate ligand and gradually creating an ‘island in the sea’ folate pattern on nanoparticle surface leads to a decreased cell uptake for all the molar substitution ratios.
The matrix in Table 1 also allows an overall comparison between cell internalisation of nanoparticles with relatively clustered ligand pattern with their counterparts prepared at similar content of folate ligand, but in a more dispersed pattern (comparison in diagonal direction of Table 1, e.g. nanoparticles surface modified by a 75/25 mixture of FA-OVA conjugate at molar substitution ratio of 13.2 with 6.7 nmol FA are compared with nanoparticles of similar folate content of 6.5 nmol, but surface modified with FA-OVA conjugate at low molar substitution ratio of 3.3). Such comparisons typically reveal that statistically higher nanoparticle internalisation by Calu-3 cells is associated with a more clustered rather than dispersed surface ligand pattern. This finding appears in line with the suggested formation of folate receptors into discrete clusters at the cell membrane associated with a caveolae structure.33,34
Selected data from Table 1 are summarised in Fig. 7, which compares the internalisation of folate-decorated nanoparticles with a similar folate content, distributed at the surface in either more clustered or disperse arrangements. The trends in the figure indicate an interesting phenomenon; at higher overall ligand content, cellular internalisation of nanoparticles is higher in the clustered ligand, relative to the dispersed series. However at the low overall ligand content the trends are reversed and the internalisation of dispersed ligand nanoparticles is higher, with a crossover at the folate content of approximately 2.6–2.9 nmol.
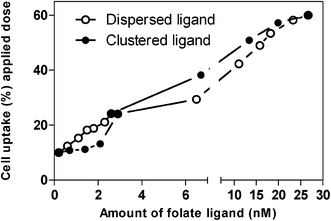 | ||
| Fig. 7 Effect of surface ligand distribution on cellular internalisation of nanoparticles at corresponding ligand amounts (selected data from Table 1). | ||
Relating our work with recently published data from other studies, it must be noted that liposomes, surface modified with either ‘individual’ or ‘clustered’ mannosyl ligands prepared with multi-branched mannosylated lipids, also displayed a higher binding affinity for the mannose receptor than vesicles containing the mono-mannosylated analogs.35 Our data are also in line with a very recent publication on the effect of folate ligand clustering, where the authors concluded that a particular ligand clustering formation at the surface of mixed micelles has a prominent effect on their association with cells in vitro and is reflected in an in vivo assessment of the system where optimal clustering was suggested to have higher association with cancer tissue.8 However there are experimental differences. The present study measures ‘real’ cellular internalisation of the nanoparticles (extracellularly present fluorescence was quenched) rather than the broader term of ‘cell associated fluorescence’ which takes into account internalised as well as cell membrane associated nanoparticles, as referred to in the above publication. Furthermore, Calu-3 cells used in our work appear to show ‘moderate’ folate expression (although further study would be need to quantify this expression), in comparison to KB cells typically used in other studies which are recognised for high overexpression of folate receptors.36,37 In summary, the work contributes to our understanding of a complex interplay of different factors involved in biological processes of cellular uptake of targeted delivery systems.
Conclusions
This work describes a simple approach to the surface modification of nanoparticle systems, which, in addition to allowing control of surface density of a ligand, also offers manipulation of ligand clustering. Our data suggests that ligand density not only affects the level of cell internalisation of targeted nanoparticles, but also influences the pathway of nanoparticle internalisation by the cells, a phenomenon not investigated previously and which determines intercellular processing of the internalised material. Ligand presentation in a clustered as opposed to dispersed arrangement affects and generally increases the level of cell uptake in an in vitro epithelial model. The system allows tuning of ligand arrangement on the surface of nanoparticles and could be a useful platform to study targeted delivery of a new generation of nano-scale therapeutics.Experimental
Preparation of folate-conjugated ovalbumin (OVA)
All chemicals were purchased from Sigma-Aldrich (UK) unless otherwise specified.OVA (40 mg) was dissolved in 0.1 M sodium carbonate buffer (pH 9.5). Folic acid (FA) was then added (in 20- to 40-fold molar excess to OVA) in combination with 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (ECDI) (in 60-fold molar excess to OVA) to OVA solution. This was expected to give a conjugate with different molar substitution ratios (MSR), ranging from 1–15 moles of FA per mole of OVA. Solution pH was maintained at pH 6.6 (by addition of HCl) during the first 15 min and then the reaction was left overnight at room temperature with continuous stirring.
The resulting conjugate was desalted and purified using a PD10 column (Sephadex G-25 Medium) (GE Healthcare, UK), with phosphate buffered saline (PBS, containing 0.14 M NaCl and 0.01 M phosphate at pH 7.4) as the eluting solution to remove any free unconjugated drug. Molar ratio of FA to OVA was determined by quantifying FA using spectrophotometry at λ364 (ε= 6.5 × 103) (Beckman DU-640 Spectrophotometer) and OVA by the Bradford reagent (using UV at 595 nm), using calibration curves of known OVA concentrations.
Preparation of folate modified polystyrene nanoparticles
FA-OVA conjugates and unconjugated OVA were adsorbed on the surface of sulphate-modified, polystyrene latex of 30 nm diameter (Green, Sigma-Aldrich, UK). To achieve this, 10 μl of nanoparticle suspension (2.5% w/v solid content) was incubated with FA-OVA conjugates, or OVA (0.2 mg in both cases) in 0.01 M PBS for three hours (at room temperature) with gentle stirring. Following incubation, nanoparticle samples were then centrifuged (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g; 30 min; using Eppendorf 5417 R centrifuge, AG 22331 Hamburg), following which the supernatant was removed and the nanoparticle pellet resuspended in Hank's Balanced Salt Solution (HBSS) (pH 7.4).
000 g; 30 min; using Eppendorf 5417 R centrifuge, AG 22331 Hamburg), following which the supernatant was removed and the nanoparticle pellet resuspended in Hank's Balanced Salt Solution (HBSS) (pH 7.4).
Light-scattering measurements
The hydrodynamic diameter and size distribution of nanoparticles was determined by Dynamic Light Scattering (DLS) (Viscotek, UK). FA-OVA and unmodified nanoparticles were analysed by DLS suspended in HBSS at pH 7.4 (under the same conditions in which nanoparticles were applied to the cells). The results represent the mean of 10 measurements.Cell culture
Calu-3 cells (used between passages 25–40) were obtained from the (American Type Culture Collection) ATCC and maintained in a 1:1 mixture of Ham's F12 (Gibco, UK) and Dulbecco's Modified Eagle Medium (DMEM) (Sigma-Aldrich, UK), supplemented with penicillin (100 units/ml), streptomycin (0.1 mg ml−1), amphotericin B (0.25 μg ml−1), Fetal Calf Serum (FCS) (10%) and L-glutamine (2 mM). Calu-3 cells were cultured to confluence in 75 cm2 flasks at 5% CO2, 37 °C. Once confluent, cells were detached from the flasks and seeded on permeable supports (Transwell®, 12 mm diameter, 0.4 μm pore size, Corning Costar, Holland) at a seeding density of 105 cells per well. The cells were bathed in 0.5 ml and 1.5 ml of folate-free RPMI 1640 medium (Sigma-Aldrich, UK) containing the above-mentioned supplements, in the apical and basolateral side, respectively. After seeding, the cells were maintained at 5% CO2, 37 °C and the culture medium changed every other day. The integrity of the cell layers and tight junction formation was assessed by measurement of Transepithelial Electrical Resistance (TEER) as detailed in our previous publications.38
Folate receptor immunostaining
Calu-3 cells were cultured as layers on permeable supports (as described above). Folate receptor immunostaining was conducted using mouse, anti-human FOLR1 primary antibody (Abnova Corporation, UK) and goat, anti-mouse TRITC-labelled secondary antibody (Sigma-Aldrich, UK). Cell layers were fixed in 4% parafomaldehyde, washed with PBS and permeabilised by incubating with Triton X-100 (0.1% v/v in PBS). Samples were then washed again with PBS and incubated for 1 h with bovine serum albumin (BSA) solution in PBS (1% w/v). Mouse, anti-human FOLR1 was diluted to 10 μg ml−1 in 1% BSA/PBS and incubated with the cells for 30–60 min at room temperature. Cells were then washed repeatedly with PBS and treated with goat, anti-mouse TRITC-labelled secondary antibody (diluted according to the supplier's instructions in 1% BSA/PBS) for 30–60 min. Finally, the samples were washed extensively with PBS (5×), permeable supports excised and mounted on glass slides using 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI)-containing, ProLong® Gold antifade mounting medium (Invitrogen, UK) for confocal imaging. Samples were imaged using a Leica TCS SP2 system mounted on a Leica DMIRE2 inverted microscope.Cell uptake studies
Before the addition of nanoparticle samples, the culture medium was removed and the Calu-3 cell layers rinsed with PBS. Cells were allowed to equilibrate for 1 h at 37 °C in HBSS, buffered with 15 mM 4-(2-hydroxyethyl)-1-piperazine-ethane sulfonic acid (HEPES, pH 7.4). OVA-FA modified and unmodified nanoparticles (0.25% w/v solid, 1.69 × 1013 nanoparticles) in HBSS were added to the apical side of the cell layers. The uptake study was performed for a period of four hours. The samples were then removed from the apical side of the cell layers and the cells washed with 0.4% (w/v) Trypan Blue solution to quench extracellular fluorescence. The cells were then digested with 0.2 N NaOH in 0.5% Triton X-100 and centrifuged at 25000 g (using Eppendorf 5417 R centrifuge, AG 22331 Hamburg) for 30 s. The fluorescence intensity of the supernatant was measured by a Fluorescence Spectrophotometer (Varian Cary Eclipse Fluorescence Spectrophotometer).
To assess the route of cell internalisation of nanoparticles, Calu-3 cells were pre-treated with either clathrin inhibitor, chlorpromazine (10 μg ml−1), or caveolae inhibitor, filipin (5 μg ml−1) for 1 h prior to the addition of nanoparticles. Inhibitors were used at concentrations that did not exhibit a prominent effect on cell viability (<15% decrease in cell viability relative to control, ESI,† Fig. S1). Nanoparticle internalisation was assessed using the method described above, but in the presence of endocytosis inhibitors.
Statistical analysis
All experiments were carried out three times using triplicate samples. One way analysis of variance (ANOVA) followed by Bonferroni post-hoc test was applied for comparison of group means at a p value of 0.05, unless otherwise stated.References
- Y. H. Shen, J. Chen, Q. F. Liu, C. C. Feng, X. L. Gao, L. Wang, Q. Z. Zhang and X. G. Jiang, Int. J. Pharm., 2011, 413, 184–193 CrossRef CAS.
- A. Fakhari, A. Baoum, T. J. Siahaan, K. B. Le and C. Berkland, J. Pharm. Sci., 2011, 100, 1045–1056 CrossRef CAS.
- F. Gu, L. Zhang, B. A. Teply, N. Mann, A. Wang, A. F. Radovic-Moreno, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 2586–2591 CrossRef CAS.
- A. Garg, A. W. Tisdale, E. Haidari and E. Kokkoli, Int. J. Pharm., 2009, 366, 201–210 CrossRef CAS.
- M. E. Gindy, S. X. Ji, T. R. Hoye, A. Z. Panagiotopoulos and R. K. Prud'homme, Biomacromolecules, 2008, 9, 2705–2711 CrossRef CAS.
- A. O. Saeed, J. P. Magnusson, E. Moradi, M. Soliman, W. Wang, S. Stolnik, K. J. Thurecht, S. M. Howdle and C. Alexander, Bioconjugate Chem., 2011, 22, 156–168 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- Z. Poon, S. Chen, A. C. Engler, H. I. Lee, E. Atas, G. von Maltzahn, S. N. Bhatia and P. T. Hammond, Angew. Chem., Int. Ed., 2010, 49, 7266–7270 CrossRef CAS.
- K. J. Kim and A. B. Malik, Am J Physiol Lung Cell Mol Physiol, 2003, 284, L247–259 CAS.
- P. E. Stein, A. G. Leslie, J. T. Finch and R. W. Carrell, J. Mol. Biol., 1991, 221, 941–959 CrossRef CAS.
- H. B. Williams and A. R. Choppin, J. Gen. Physiol., 1950, 34, 183–192 CrossRef CAS.
- A. V. Elgersma, R. L. J. Zsom, W. Norde and J. Lyklema, J. Colloid Interface Sci., 1990, 138, 145–156 CrossRef CAS.
- B. D. Fair and A. M. Jamieson, J. Colloid Interface Sci., 1980, 77, 525–534 CrossRef CAS.
- A. Kondo and K. Higashitani, J. Colloid Interface Sci., 1992, 150, 344–351 CrossRef CAS.
- J. Revilla, A. Elaissari, P. Carriere and C. Pichot, J. Colloid Interface Sci., 1996, 180, 405–412 CrossRef CAS.
- N. Parker, M. J. Turk, E. Westrick, J. D. Lewis, P. S. Low and C. P. Leamon, Anal. Biochem., 2005, 338, 284–293 CrossRef CAS.
- M. D. Salazar and M. Ratnam, Cancer Metastasis Rev., 2007, 26, 141–152 CrossRef CAS.
- M. O. Oyewumi and R. J. Mumper, Int. J. Pharm., 2003, 251, 85–97 CrossRef CAS.
- G. F. Dawson and G. W. Halbert, Pharm. Res., 2000, 17, 1420–1425 CrossRef CAS.
- C. M. Paulos, J. A. Reddy, C. P. Leamon, M. J. Turk and P. S. Low, Mol. Pharmacol., 2004, 66, 1406–1414 CrossRef CAS.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS.
- R. G. Anderson and K. Jacobson, Science, 2002, 296, 1821–1825 CrossRef CAS.
- N. P. Gabrielson and D. W. Pack, J. Controlled Release, 2009, 136, 54–61 CrossRef CAS.
- K. M. Kitchens, R. B. Kolhatkar, P. W. Swaan and H. Ghandehari, Mol. Pharmaceutics, 2008, 5, 364–369 CrossRef CAS.
- F. M. Brodsky, Molecular Biology of the Cell, 2001, 12, 1A–1A Search PubMed.
- J. Rejman, A. Bragonzi and M. Conese, Mol. Ther., 2005, 12, 468–474 CrossRef CAS.
- E. J. Smart, C. Mineo and R. G. Anderson, J. Cell Biol., 1996, 134, 1169–1177 CrossRef CAS.
- S. Sabharanjak and S. Mayor, Adv. Drug Delivery Rev., 2004, 56, 1099–1109 CrossRef CAS.
- J. S. Rodman, L. Seidman and M. G. Farquhar, J. Cell Biol., 1986, 102, 77–87 CrossRef CAS.
- H. Birn, J. Selhub and E. I. Christensen, American Journal of Physiology, 1993, 264, C302–C310 CAS.
- S. Stolnik, B. Daudali, A. Arien, J. Whetstone, C. R. Heald, M. C. Garnett, S. S. Davis and L. Illum, Biochim. Biophys. Acta, Biomembr., 2001, 1514, 261–279 CrossRef CAS.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS.
- K. G. Rothberg, Y. S. Ying, B. A. Kamen and R. G. Anderson, J. Cell Biol., 1990, 111, 2931–2938 CrossRef CAS.
- M. Wu, J. Fan, W. Gunning and M. Ratnam, J. Membr. Biol., 1997, 159, 137–147 CrossRef CAS.
- B. Frisch, S. Espuelas, C. Thumann, B. Heurtault and F. Schuber, Bioconjugate Chem., 2008, 19, 2385–2393 CrossRef.
- V. Dixit, J. Van den Bossche, D. M. Sherman, D. H. Thompson and R. P. Andres, Bioconjugate Chem., 2006, 17, 603–609 CrossRef CAS.
- S. D. Weitman, R. H. Lark, L. R. Coney, D. W. Fort, V. Frasca, V. R. Zurawski and B. A. Kamen, Cancer Res, 1992, 52, 3396–3401 CAS.
- E. Moradi, G. Byrne, M. C. Garnett, F. H. Falcone and S. Stolnik, presented in part at the Controlled Release society, Portlan, USA, 2010 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra01168a/ |
| This journal is © The Royal Society of Chemistry 2012 |