DOI:
10.1039/C2RA01235A
(Paper)
RSC Adv., 2012,
2, 3451-3457
Magnesium complexes supported by pyrrolyl ligands: syntheses, characterizations, and catalytic activities towards the polymerization of ε-caprolactone†
Received
4th December 2011
, Accepted 20th January 2012
First published on 29th February 2012
Abstract
The syntheses, structures and catalytic activities of five magnesium complexes supported by HL1 (HL1 = N-(1H-pyrrol-2-ylmethylene)(2-pyridinyl)methanimine), HL2 (HL2 = N-(1H-pyrrol-2-ylmethylene)-2-pyridineethanimine), HL3 (HL3 = 2-pyrrolecarbaldmethylimine), HL4 (HL4 = N-((1H-pyrrol-2-yl)methylene)(phenyl)methanimine) and HL5 (HL5 = N-((1H-pyrrol-2-yl)methylene)-2-phenylethanimine) are described. Treatment of MgnBu2 with 2 equiv. of HL1, HL2, HL3, HL4 and HL5, respectively, results in the formation of Mg(L1)2 (1), Mg(L2)2 (2), Mg(L3)2(THF)2 (3), Mg(L4)2(THF)2 (4) and Mg(L5)2(THF)2 (5). All complexes have been characterized by elemental analyses and NMR studies. The solid-state structures of complexes 2, 3, and 4 have been further established by single X-ray crystallography. All complexes were found to be active catalysts for the ring-opening polymerization of ε-caprolactone.
Introduction
Magnesium compounds are among the most common organometallic reagents used as indispensable tools in organic synthesis and catalysis,1 especially as alkylating reagents,2 catalysts for the hydroamination of aminoalkenes,3 and initiators for the polymerization of esters.4 As new applications of magnesium compounds continue to be discovered, studies tailoring ligand systems to specific utilizations attract increasing attention. Over the past few years, the exploitation of magnesium compounds supported by bulky β-diketiminato ligand systems has provided a number of spectacular results,5 and a plethora of magnesium alkoxides supported by such ligands have been found as initiators for ring-opening polymerization of organic esters.6 However, the potential of alkylmagnesium complexes supported by pyrrolyl ligands, especially the pyrrolyl Schiff base ligands that form five-membered ring systems with magnesium ions, to be the catalysts for hydroamination or polymerization reactions has been less explored.7 The deprotonated pyrrole, named the pyrrolide anion, is among the most favorable ligands in organometallic chemistry. Isoelectronic with the Cp anion, the pyrrolide anion is also a competent η5 ligand and can offer the possibility of forming o-bonds through the ring nitrogen atom. Thus, pyrrolyl ligands provide tunable steric and electronic features required for compensating coordinative unsaturation of metal centers and catalytic activity toward polymerization. It is reasoned that the reaction of MgnBu2 with bi- or tridentate pyrrolyl Schiff base ligands could provide alkylmagnesium complexes, which may show good catalytic activities towards hydroamination and polymerization reactions. Consequently, we have explored the reactions of MgnBu2 with two tridentate ligands HL1 and HL2, and three bidentate ligands HL3–HL5 (Scheme 1), and five complexes of compositions Mg(L1)2 (1), Mg(L2)2 (2), Mg(L3)2(THF)2 (3), Mg(L4)2(THF)2 (4) and Mg(L5)2(THF)2 (5) were generated. Herein, we report the syntheses and characterizations of these complexes. The catalytic behavior of these five complexes towards ring-opening polymerization of ε-caprolactone is also presented.
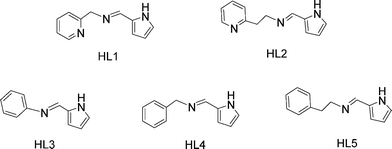 |
| | Scheme 1 Structures of the ligands. | |
Results and discussion
Syntheses of ligands and magnesium complexes
The ligands HL1 (HL1 = N-(1H-pyrrol-2-ylmethylene)(2-pyridinyl)methanimine),8 HL2 (HL2 = N-(1H-pyrrol-2-ylmethylene)-2-pyridineethanimine),9 HL3 (HL3 = 2-pyrrolecarbaldmethylimine),10 HL4 (HL4 = N-((1H-pyrrol-2-yl)methylene)(phenyl) methanimine),11 and HL5 (HL5 = N-((1H-pyrrol-2-yl)methylene)-2-phenyl ethanimine)11 were prepared using condensation reactions between pyrrole-2-carboxaldehyde with the corresponding amine. Treatment of MgnBu2 with one equiv. of HL1, HL2, HL3, HL4 and HL5, respectively, led to the formations of 1–5. The one ligand set chelated alkylmagnesium complex was not afforded. Then the reaction of MgnBu2 with 2 equiv. of HL1, HL2, HL3, HL4 and HL5, respectively, was conducted, and complexes 1–5 were readily afforded in good yields after recrystallization in THF/hexane (Scheme 2). They were also characterized by 1H and 13C NMR spectra and elemental analyses.
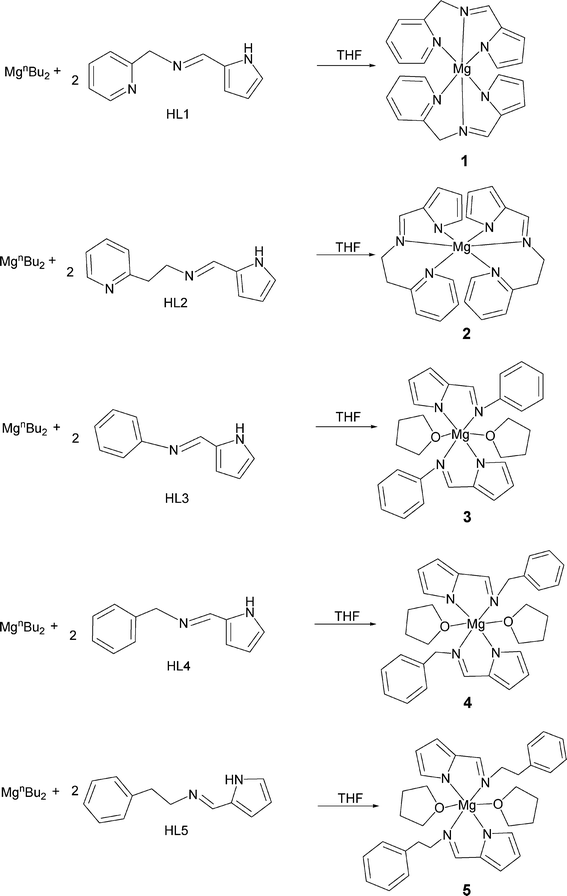 |
| | Scheme 2 Syntheses of complexes 1–5. | |
Structure descriptions of complexes 2, 3 and 4
The molecular structures of 2, 3 and 4 in the solid states have been confirmed by X-ray analysis and are shown in Fig. 1, 2, and 3, respectively. The crystallographic data and experimental details for structural analyses are summarized in Table 1. Selected bond distances and angles are listed in Table 2.
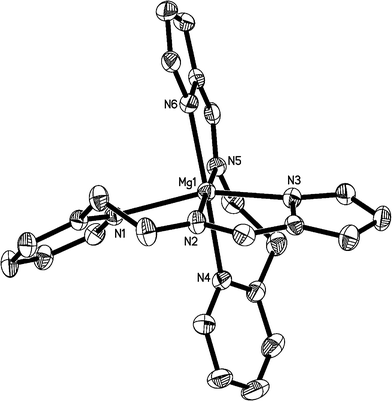 |
| | Fig. 1 ORTEP structural drawing of 2. Ellipsoids are drawn at the 30% probability level, and hydrogen atoms are omitted for clarity. | |
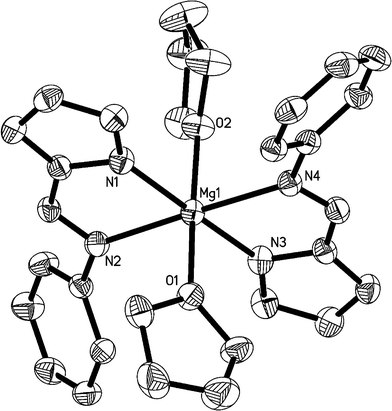 |
| | Fig. 2 ORTEP structural drawing of 3. Ellipsoids are drawn at the 30% probability level, and hydrogen atoms are omitted for clarity. | |
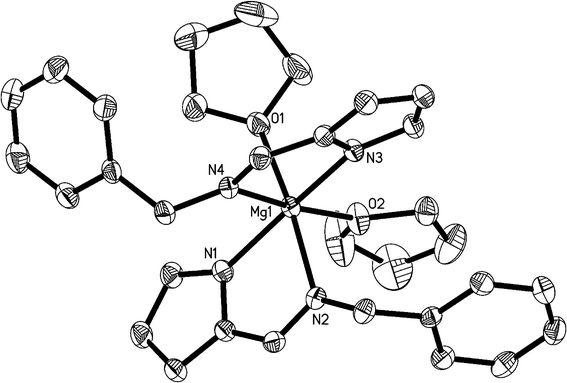 |
| | Fig. 3 ORTEP structural drawing of 4. Ellipsoids are drawn at the 30% probability level, and hydrogen atoms are omitted for clarity. | |
| |
2
|
3
|
4
|
|
Including solvate molecules.
Mo-Kα radiation.
R
1 = Σ(|Fo | − |Fc|)/Σ(|Fo|) for observed reflections.
w = 1/[σ2(Fo2) + (αP)2 + bP] and P = [max(Fo2,0) + 2Fc2]/3.
wR
2 = {Σ[w(Fo2 − Fc2)2]/Σ[w(Fo2)2]}1/2 for all data.
|
| Formulaa |
C24H24MgN6 |
C30H34 MgN4O2 |
C32H38 MgN4O2 |
| M g mol−1a |
420.80 |
506.92 |
534.97 |
|
T/K |
293(2) |
293(2) |
293(2) |
| Wavelengthb/Å |
0.71073 |
0.71073 |
0.71073 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
C 2/c |
P 21/c |
Pī |
|
a/Å |
28.215(6) |
9.986(2) |
10.453(2) |
|
b/Å |
16.504(3) |
14.269(3) |
11.456(2) |
|
c/Å |
14.405(3) |
19.278(4) |
12.516(3) |
|
α/° |
90 |
90 |
84.02(3) |
|
β/° |
98.59(3) |
92.42(3) |
76.88(3) |
|
γ/° |
90 |
90 |
89.43(3) |
|
V/Å3 |
6633(2) |
2744.5(10) |
1451.7(5) |
|
Z
|
12 |
4 |
2 |
|
ρ/g cm−3 |
1.264 |
1.227 |
1.224 |
|
F(000) |
2664 |
1080 |
572 |
| Crystal size/mm |
0.40 × 0.30 × 0.15 |
0.23 × 0.21 × 0.19 |
0.25 × 0.20 × 0.17 |
|
θ range/° |
3.05 to 25.02° |
3.04 to 27.47° |
1.68 to 28.36° |
| Limiting indices |
−29 ≤ h ≤ 33 |
−12 ≤ h ≤ 12 |
−13 ≤ h ≤ 13 |
| −19 ≤ k ≤ 18 |
−18 ≤ k ≤ 16 |
−15 ≤ k ≤ 15 |
| −17 ≤ l ≤ 16 |
−25 ≤ l ≤ 20 |
−16 ≤ l ≤ 15 |
| Reflections collected/unique |
16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 422/5828 422/5828 |
14835/6228 |
26473/7247 |
| Data/restraints/parameters |
5828/0/421 |
6228/7/334 |
7247/15/352 |
| GOF |
1.181 |
1.157 |
1.066 |
|
R
1, wR2 [I > 2σ(I)] |
R
1 = 0.0927 |
R
1 = 0.0980 |
R
1 = 0.0556 |
|
wR
2 = 0.1372 |
wR
2 = 0.2011 |
wR
2 = 0.1497 |
|
R
1
c,wR2d,e (all data) |
R
1 = 0.1359 |
R
1 = 0.1504 |
R
1 = 0.0685 |
|
wR
2 = 0.1537 |
wR
2 = 0.2289 |
wR
2 = 0.1597 |
| Largest diff. peak and hole/e Å3 |
0.284 and −0.254 |
0.488 and −0.736 |
0.519 and −0.501 |
Table 2 Selected bond lengths (Å) and angles (°) for 2–4
|
2
|
| Mg(1)-N(1) |
2.307(4) |
Mg(1)-N(4) |
2.320(4) |
| Mg(1)-N(2) |
2.176(3) |
Mg(1)-N(5) |
2.162(3) |
| Mg(1)-N(3) |
2.169(4) |
Mg(1)-N(6) |
2.156(4) |
| N(1)-Mg(1)-N(2) |
82.67(13) |
N(2)-Mg(1)-N(6) |
105.47(13) |
| N(1)-Mg(1)-N(3) |
156.89(14) |
N(3)-Mg(1)-N(4) |
84.44(13) |
| N(1)-Mg(1)-N(4) |
84.47(12) |
N(3)-Mg(1)-N(5) |
101.98(13) |
| N(1)-Mg(1)-N(5) |
96.59(13) |
N(3)-Mg(1)-N(6) |
103.93(14) |
| N(1)-Mg(1)-N(6) |
93.27(13) |
N(4)-Mg(1)-N(5) |
82.49(13) |
| N(2)-Mg(1)-N(3) |
77.97(13) |
N(4)-Mg(1)-N(6) |
159.88(13) |
| N(2)-Mg(1)-N(4) |
94.09(13) |
N(5)-Mg(1)-N(6) |
77.90(13) |
| N(2)-Mg(1)-N(5) |
176.56(15) |
|
|
|
3
|
| Mg(1)-N(1) |
2.144(3) |
Mg(1)-N(4) |
2.257(3) |
| Mg(1)-N(2) |
2.261(3) |
Mg(1)-O(1) |
2.121(3) |
| Mg(1)-N(3) |
2.125(3) |
Mg(1)-O(2) |
2.141(3) |
| N(1)-Mg(1)-O(1) |
92.82(11) |
N(2)-Mg(1)-N(4) |
178.19(11) |
| N(1)-Mg(1)-O(2) |
87.46(11) |
N(3)-Mg(1)-O(1) |
90.74(11) |
| N(1)-Mg(1)-N(2) |
78.49(11) |
N(3)-Mg(1)-O(2) |
89.06(11) |
| N(1)-Mg(1)-N(3) |
175.95(12) |
N(3)-Mg(1)-N(4) |
78.67(11) |
| N(1)-Mg(1)-N(4) |
103.31(11) |
N(4)-Mg(1)-O(1) |
89.02(11) |
| N(2)-Mg(1)-O(1) |
90.76(11) |
N(4)-Mg(1)-O(2) |
88.81(11) |
| N(2)-Mg(1)-O(2) |
91.41(11) |
O(1)-Mg(1)-O(2) |
177.82(12) |
| N(2)-Mg(1)-N(3) |
99.53(12) |
|
|
|
4
|
| Mg(1)-N(1) |
2.1393(15) |
Mg(1)-N(4) |
2.2124(16) |
| Mg(1)-N(2) |
2.1938(17) |
Mg(1)-O(1) |
2.1406(16) |
| Mg(1)-N(3) |
2.1480(15) |
Mg(1)-O(2) |
2.1455(15) |
| N(1)-Mg(1)-O(1) |
94.63(6) |
N(2)-Mg(1)-N(4) |
86.95(6) |
| N(1)-Mg(1)-O(2) |
93.07(6) |
N(3)-Mg(1)-O(1) |
92.66(7) |
| N(1)-Mg(1)-N(2) |
78.65(6) |
N(3)-Mg(1)-O(2) |
95.30(6) |
| N(1)-Mg(1)-N(3) |
169.32(6) |
N(3)-Mg(1)-N(4) |
78.74(6) |
| N(1)-Mg(1)-N(4) |
92.95(6) |
N(4)-Mg(1)-O(1) |
94.17(6) |
| N(2)-Mg(1)-O(1) |
173.25(6) |
N(4)-Mg(1)-O(2) |
173.98(6) |
| N(2)-Mg(1)-O(2) |
94.34(6) |
O(1)-Mg(1)-O(2) |
85.23(7) |
| N(2)-Mg(1)-N(3) |
94.09(6) |
|
|
Single crystal analysis revealed that complex 2 crystallizes in monoclinic crystal system of the C2/c space group. The central MgII ion possesses a distorted octahedral coordination environment with six nitrogen atoms from two tridentate L1− ligands. Two donor imine nitrogen atoms of the ligands are in trans arrangement, with bond angles of 77.97(13)° (N2–Mg1–N3) and 101.98(13)° (N3–Mg1–N5), summing to 179.97(6)°. The atoms N1, N3, N4 and N6 are almost coplanar, with the bond angles of 103.93(14)° (N3–Mg1–N6), 84.44(13)° (N3–Mg1–N4), 84.47 (12)° (N1–Mg1–N4) and 93.27(13)° (N1–Mg1–N6), respectively, summing to 366.16(6)°, with the deviation being 6.16(6)° compared with 360°. The bond lengths between the magnesium atom and donor pyridyl nitrogen atom (Mg1–N1 = 2.307(4) Å, and Mg1–N4 = 2.320(4) Å) are apparently longer than those of the Mg–N(imine) distances (Mg1–N2 = 2.176(3) Å, and Mg1–N5 = 2.162(3) Å), and also longer than the Mg–N(pyrrolyl) distances (Mg1–N3 = 2.169(4) Å, and Mg1–N6 = 2.156(4) Å). Compared with the four-coordinated magnesium complexes [Mg–N(imine) = 2.0525(12) and Mg–N(pyrrolyl) = 2.1154(13)]7a,7b, the Mg–N(imine) and Mg–N(pyrrolyl) distances in 2 are all longer.
The crystal structure of 3 revealed that the octahedral MgII ion is surrounded by four nitrogen atoms (N1, N2, N3, and N4) from two chelating pyrrolyl ligands and two oxygen atoms (O1 and O2) from two THF molecules, with two oxygen atoms occupying the axial positions. The atoms N1, N2, N3 and N4 form the equatorial plane, with bond angles of 78.49(11)° (N1–Mg1–N2), 99.53(12)° (N3–Mg1–N2), 78.67(11)° (N3–Mg1–N4) and 103.31(11)° (N1–Mg1–N4), respectively, summing to 360.04(5)°, with the deviation being 0.04(5)° compared with 360°. The bond lengths between the magnesium atom and donor nitrogen atoms (Mg1–N2 = 2.261(3) Å, Mg1–N4 = 2.257(3) Å) are apparently longer than the Mg–N(pyrrolyl) distance (Mg1–N1 = 2.144(3) Å , Mg1–N3 = 2.125(3) Å). The two THF ligands are trans to each other, as are the two imines and the two pyrrolyl ligands.
The crystal structure of 4 is similar to that of 3, and the metal ion is also coordinated by two ligands and two THF molecules. The two THF ligands and the two imines are cis to each other, while two pyrrolyl ligands are trans to each other in complex 4.
The application of structurally well-defined magnesium complexes as initiators for the synthesis of poly(ε-caprolactone) (PCL) via the ring-opening polymerization of lactones has gained particular interest.4,6 Among the systems evaluated are phenolate-,12β-diiminato-,5a,5c,6a,6b and trisindazalylborate13 ligand supported magnesium complexes. To the best of our knowledge, the catalytic activity of magnesium complexes chelated by pyrrolyl Schiff base ligands has not been studied and this represents a deficiency in this area. Therefore, we carried out an assessment of the ring-opening polymerization capability of the five new complexes.
The initial studies were performed using Mg(L1)2 (1) as the initiator and employing dimethyl ether (DME), tetrahydrofuran (THF), and toluene as the solvent, respectively. It was found that no observable polymerization occurred at 20 °C in each solvent after 24 h. As the temperature was increased to 40 °C, trace amounts of polymers were generated. A good yield (90%) was afforded in toluene, a moderate yield (48%) was provided in DME and a poor yield (23%) was given in THF as the temperature was further increased to 60 °C. When the polymerization was conducted at 80 °C, the comparable yields could be achieved in a shorter time.
We further probed the polymerization activity of complexes 2–5 at 60 °C and 80 °C in DME, THF and toluene, respectively. The polymerization results are summarized in Table 3. It was found that all five complexes can effectively initiate ε-caprolactone polymerization, and all of the obtained polymers have high molecular weights and relatively narrow molecular weight distributions (PDIs). The solvents have the obvious effect on the yields of the polymers. For complexes 1 and 2, when the polymerization was conducted in ethereal solvents, such as DME and THF, poor or moderate yields were afforded; whereas very good yields were obtained when the polymerizations were performed in toluene. We reasoned that DME and THF molecules tend to coordinate with the metal center during the polymerization process, which may hinder the attack of ε-caprolactone to the same metal center. No remarkable differences in different solvent were observed for the polymerization reactions catalyzed by complexes 3–5.
| Entry |
Initiator |
Solvent |
[M/I] |
T/h |
Yielda /% |
T/°C |
Mnb (calc) (104) |
Mnc (104) |
Mnd (104) |
PDI |
Efficiency/% |
|
Yield: weight of polymer obtained/weight of monomer used.
Mn (calc) = Mmono × [M]/[I] × Conv.
Measured by GPC relative to polystyrene standards.
Measured by GPC relative to polystyrene standards with Mark-Houwink corrections14 for Mn (obsd) = 0.56 Mn (GPC) for ε-caprolactone.
|
| 1 |
1
|
DME |
200 |
21 |
48 |
60 |
1.09 |
1.59 |
0.89 |
1.47 |
69.9 |
| 2 |
1
|
DME |
200 |
5 |
15 |
80 |
0.34 |
0.55 |
0.31 |
1.25 |
61.8 |
| 3 |
1
|
THF |
200 |
8 |
23 |
60 |
0.52 |
0.69 |
0.39 |
1.11 |
75.4 |
| 4 |
1
|
THF |
200 |
3 |
18 |
80 |
0.41 |
0.54 |
0.30 |
1.16 |
75.9 |
| 5 |
1
|
Tol |
200 |
20 |
90 |
60 |
2.05 |
3.18 |
1.78 |
1.78 |
64.5 |
| 6 |
1
|
Tol |
200 |
10 |
92 |
80 |
2.10 |
3.01 |
1.68 |
1.77 |
69.7 |
| 7 |
2
|
DME |
200 |
10 |
23 |
60 |
0.52 |
0.72 |
0.40 |
1.21 |
72.2 |
| 8 |
2
|
DME |
200 |
2 |
20 |
80 |
0.45 |
0.58 |
0.32 |
1.18 |
77.6 |
| 9 |
2
|
THF |
200 |
8 |
32 |
60 |
0.73 |
1.03 |
0.58 |
1.30 |
70.9 |
| 10 |
2
|
THF |
200 |
3 |
90 |
80 |
2.05 |
5.38 |
3.01 |
1.29 |
38.1 |
| 11 |
2
|
Tol |
200 |
20 |
89 |
60 |
2.03 |
9.87 |
5.53 |
1.28 |
20.6 |
| 12 |
2
|
Tol |
200 |
12 |
95 |
80 |
2.17 |
5.41 |
3.03 |
1.60 |
40.1 |
| 13 |
3
|
DME |
200 |
3 |
90 |
60 |
2.05 |
5.55 |
3.11 |
1.27 |
36.9 |
| 14 |
3
|
DME |
200 |
1 |
93 |
80 |
2.12 |
9.25 |
5.18 |
1.44 |
22.9 |
| 15 |
3
|
THF |
200 |
4 |
49 |
60 |
1.12 |
1.25 |
0.70 |
1.29 |
89.6 |
| 16 |
3
|
THF |
200 |
2 |
23 |
80 |
0.52 |
0.69 |
0.39 |
1.21 |
75.4 |
| 17 |
3
|
Tol |
200 |
5 |
50 |
60 |
1.14 |
6.20 |
3.47 |
1.63 |
18.4 |
| 18 |
3
|
Tol |
200 |
1 |
52 |
80 |
1.19 |
1.60 |
0.90 |
1.41 |
74.4 |
| 19 |
4
|
DME |
200 |
2 |
72 |
60 |
1.64 |
2.92 |
1.63 |
1.46 |
56.2 |
| 20 |
4
|
DME |
200 |
0.5 |
85 |
80 |
1.94 |
3.28 |
1.84 |
1.30 |
59.1 |
| 21 |
4
|
THF |
200 |
1.5 |
90 |
60 |
2.05 |
2.88 |
1.61 |
1.36 |
71.2 |
| 22 |
4
|
THF |
200 |
0.5 |
76 |
80 |
1.73 |
2.56 |
1.43 |
1.34 |
67.6 |
| 23 |
4
|
Tol |
200 |
1 |
86 |
60 |
1.96 |
2.31 |
1.29 |
1.43 |
84.8 |
| 24 |
4
|
Tol |
200 |
0.4 |
68 |
80 |
1.55 |
2.33 |
1.30 |
1.43 |
66.5 |
| 25 |
5
|
DME |
200 |
6 |
28 |
60 |
0.64 |
0.72 |
0.40 |
1.22 |
88.9 |
| 26 |
5
|
DME |
200 |
3 |
23 |
80 |
0.52 |
0.65 |
0.36 |
1.20 |
80.7 |
| 27 |
5
|
THF |
200 |
6 |
85 |
60 |
1.94 |
3.82 |
2.14 |
1.51 |
50.8 |
| 28 |
5
|
THF |
200 |
3 |
31 |
80 |
0.71 |
0.91 |
0.51 |
1.20 |
78.1 |
| 29 |
5
|
Tol |
200 |
2 |
75 |
60 |
1.71 |
2.61 |
1.46 |
1.50 |
65.5 |
| 30 |
5
|
Tol |
200 |
1 |
32 |
80 |
0.73 |
1.10 |
0.62 |
1.42 |
66.4 |
Conclusions
In conclusion, five new complexes Mg(L1)2 (1), Mg(L2)2 (2), Mg(L3)2(THF)2 (3), Mg(L4)2(THF)2 (4) and Mg(L5)2(THF)2 (5) were synthesized and characterized. The catalytic behavior of these complexes towards ring-opening polymerization of ε-caprolactone in different solvents and at different temperatures was investigated. Complexes 1–5 exhibited good catalytic activity for the polymerization of ε-caprolactone.
Experimental section
General considerations
All manipulations of air-sensitive complexes were carried out in a MBraun drybox under a purified nitrogen atmosphere. Anhydrous THF, DME and toluene were freshly distilled from purple sodium benzophenone ketyl for at least 4 days. 1H and 13C spectra were recorded on Innova-400 and Unity Innova-300 spectrometers at ambient temperature using TMS as an internal standard, and chemical shifts were reported in ppm. Elemental analyses for carbon, hydrogen and nitrogen atoms were performed on a Carlo-Erba EA1110 CHNO-S microanalyzer.
X-ray crystallography
Crystals grown from concentrated solutions at room temperature were quickly selected and mounted on a glass fiber in wax. The data collections were carried out on a Mercury CCD detector equipped with graphite-monochromated Mo-Kα radiation using the ϕ/ω scan technique at room temperature. The structures were solved by direct methods with SHELXS-97.15,16 The hydrogen atoms were assigned with common isotropic displacement factors and included in the final refinement by use of geometrical restraints. A full-matrix least-squares refinement on F2 was carried out using SHELXL-97.
General procedure for the polymerization reaction
A THF solution of complex 2 (0.0211 g, 0.05 mmol) and ε-caprolactone (1.14 g, 10 mmol) was added to a 100 mL Schlenk flask equipped with a magnetic stirring bar in a glovebox. Afterwards, the Schlenk flask was taken out of the glovebox. The reaction mixture was heated to 60 °C for 8 h and then terminated by addition of a mixture of conc. HCl/EtOH (1:5 v/v) (2 mL). Petroleum ether (20 mL) was added to give a yellow solid. The solid was dissolved in THF (20 mL), and column chromatography on Al2O3 gave a white solid.
Syntheses of complexes
Mg(L1)2 (1).
To a solution of MgnBu2 (2.0 mL, 0.5 M in heptane, 1.0 mmol) in tetrahydrofuran (2 mL) was added HL1 (0.3702 g, 2.0 mmol) in tetrahydrofuran at −35 °C. After stirring at room temperature for 24 h, the volatiles were removed under reduced pressure to give a purple solid. Yield: 0.3429 g (87.32%). 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 2H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 7.93 (d, 2H, J = 4.7 Hz, pyridyl-H), 7.56 (t, 2H, J = 7.5 Hz, pyridyl-H), 7.21 (d, 2H, J = 7.8 Hz, pyridyl-H), 6.99–6.91 (m, 2H, pyridyl-H), 6.81 (s, 2H, pyrrole-H), 6.69 (d, J = 3.0 Hz, 2H, pyrrole-H), 6.11 (d, J = 1.8 Hz, 2H, pyrrole-H), 5.01 (s, 4H, NCH2). 13C NMR (101 MHz, CDCl3) δ 158.81, 158.48, 147.57, 138.34, 137.46, 136.14, 122.67, 122.00, 115.41, 110.27, 55.25. Anal. calc. for C22H20N6Mg: C, 67.28; H, 5.13; N 21.40%. Found: C, 67.98; H, 5.51; N, 20.78%.
CH), 7.93 (d, 2H, J = 4.7 Hz, pyridyl-H), 7.56 (t, 2H, J = 7.5 Hz, pyridyl-H), 7.21 (d, 2H, J = 7.8 Hz, pyridyl-H), 6.99–6.91 (m, 2H, pyridyl-H), 6.81 (s, 2H, pyrrole-H), 6.69 (d, J = 3.0 Hz, 2H, pyrrole-H), 6.11 (d, J = 1.8 Hz, 2H, pyrrole-H), 5.01 (s, 4H, NCH2). 13C NMR (101 MHz, CDCl3) δ 158.81, 158.48, 147.57, 138.34, 137.46, 136.14, 122.67, 122.00, 115.41, 110.27, 55.25. Anal. calc. for C22H20N6Mg: C, 67.28; H, 5.13; N 21.40%. Found: C, 67.98; H, 5.51; N, 20.78%.
Mg(L2)2 (2).
To a solution of MgnBu2 (2.0 mL, 0.5 M in heptane, 1.0 mmol) in tetrahydrofuran (2 mL) was added HL2 (0.3985 g, 2.0 mmol) in tetrahydrofuran at −35 °C. The reaction mixture was stirred at room temperature overnight after which time volatiles were removed under reduced pressure to yield a crude product. The final product was obtained as light yellow block crystals after crystallization from tetrahydrofuran/hexane. Yield: 0.3778 g (89.78%). 1H NMR (400 MHz, CDCl3) δ 8.15 (s, 2H, NCH), 7.99 (d, 2H, J = 4.4 Hz, pyridyl-H), 7.53 (td, J = 7.6, 1.6 Hz, 2H, pyridyl-H), 7.16 (d, J = 7.7 Hz, 2H, pyridyl-H), 6.95–6.84 (m, 4H, pyridyl-H+pyrrole-H), 6.55 (d, J = 2.8 Hz, 2H, pyrrole-H), 6.15–6.04 (m, 2H, pyrrole-H), 4.14–4.02 (m, 4H, CH2-pyridyl), 3.76 (s, 4H, NCH2). 13C NMR (101 MHz, CDCl3) δ 159.68, 152.77, 149.36, 136.22, 130.00, 123.62, 122.46, 121.29, 114.78, 109.62, 60.25, 39.95. Anal. calc. for C24H24N6Mg: C, 68.50; H, 5.75; N, 19.97%. Found: C, 68.73; H, 5.69; N, 19.59%.
Mg(L3)2(THF)2 (3).
Following a procedure similar to that described for the preparation of 1, treatment of tetrahydrofuran (2 mL) and MgnBu2 (2.0 mL, 0.5 M in heptane, 1.0 mmol) with 2 equiv. of HL3 (0.3404 g, 2.0 mmol) in tetrahydrofuran (5 mL) at −35 °C was carried out. The reaction mixture was stirred at room temperature overnight after which time volatiles were removed under reduced pressure. The product was obtained as light yellow block crystal after crystallization from tetrahydrofuran/hexane. Yield: 0.4326 g (85.35%). 1H NMR (400 MHz, CDCl3) δ 8.43 (s, 2H, NCH), 7.36 (dd, J = 15.5, 8H, Ar-H), 7.19 (t, J = 7.0 Hz, 2H, Ar-H), 6.87 (s, 2H, pyrrole-H), 6.78 (s, 2H, pyrrole-H), 6.27 (s, 2H, pyrrole-H), 3.77 (s, 8H, CH2O of THF), 1.81 (s, 8H, CH2 of THF).13C NMR (75 MHz, CDCl3) δ 155.70, 149.40, 138.70, 138.40, 129.56, 125.08, 120.94, 120.13, 113.01, 69.22, 25.28. Anal. calc. for C30H34N4O2Mg: C, 71.08; H, 6.76; N, 11.05%. Found: C, 71.17; H, 6.63; N, 11.44%.
Mg(L4)2(THF)2 (4).
Following a procedure similar to that described for the preparation of 1, treatment of tetrahydrofuran (2 mL) and MgnBu2 (2.0 mL, 0.5 M in heptane, 1.0 mmol) with 2 equiv. of HL4 (0.3684 g, 2.0 mmol) in tetrahydrofuran (5 mL) at −35 °C was carried out. The reaction mixture was stirred at room temperature overnight after which time volatiles were removed under reduced pressure. The product crystallized from tetrahydrofuran/hexane as a colorless block crystal. Yield: 0.4765 g (89.07%). 1H NMR (400 MHz, CDCl3) δ 8.24 (s, 2H, NCH), 7.53–7.41 (m, 6H, Ar-H), 7.36 (d, J = 7.3 Hz, 4H, Ar-H), 7.19 (s, 2H, pyrrole-H), 6.89 (s, 2H, pyrrole-H), 6.52 (d, J = 1.5 Hz, 2H, pyrrole-H), 4.72 (s, 4H, NCH2), 3.70 (s, 8H, CH2O of THF), 1.85 (s, 8H, CH2 of THF). 13C NMR (100 MHz, CDCl3) δ 161.16, 139.60, 137.11, 135.08, 128.39, 128.28, 127.03, 116.19, 110.80, 68.39, 60.61, 25.24. Anal. calc. for C32H38N4O2Mg: C, 71.84; H, 7.16; N, 10.47%. Found: C, 71.35; H, 6.75; N, 10.94%.
Mg(L5)2(THF)2 (5).
This complex was prepared as a red solid from reaction of HL5 (0.3965 g, 2.0 mmol) in tetrahydrofuran (5 mL) with MgnBu2 (2.0 mL, 0.5 M in heptane, 1.0 mmol) in tetrahydrofuran (2 mL) at −35 °C and recrystallization from mixed solvents of tetrahydrofuran and hexane by a similar procedure as in the synthesis of 1. Yield: 0.4913 g (87.26%). 1H NMR (300 MHz, CDCl3) δ 8.03 (s, 2H, NCH), 7.37–7.15 (s, 8H, Ar-H), 7.15–7.05 (s, 4H, Ar-H+pyrrole-H), 6.73 (s, 2H, pyrrole-H), 6.44 (s, 2H, pyrrole-H), 3.70 (s, 8H, CH2O of THF), 3.60 (s, 4H, CH2-Ar, 2.67 (s, 4H, NCH2), 1.85 (s, 8H, CH2 of THF). 13C NMR (75 MHz, CDCl3) δ 160.41, 139.81, 137.27, 134.43, 129.09, 128.50, 126.21, 115.84, 110.84, 68.71, 59.71, 38.40, 25.50. Anal. calc. for C34H42N4O2Mg: C, 72.53; H, 7.52; N, 9.95%. Found: C, 72.87; H, 6.81; N, 9.90%.
Acknowledgements
The authors appreciate the financial support of the Hundreds of Talents Program (2005012) of CAS, the Natural Science Foundation of China (20872105), the “Qinglan Project” of Jiangsu Province (Bu109805) and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
References
-
(a) For selected examples see: S. J. Bonyhasy, S. P. Green, C. Jones, S. Nembenna and A. Stasch, Angew. Chem., Int. Ed., 2009, 48, 2973 CrossRef;
(b) L. F. Sanchez-Barba, D. L. Hughes, S. M. Humphrey and M. Bochmann, Organometallics, 2006, 25, 1012 CrossRef CAS;
(c) B. J. O'Keefe, M. A. Hillmayer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC;
(d) K. B. Starowieyski, J. Lewinski, R. Wozniak, J. Lipkowski and A. Chrost, Organometallics, 2003, 22, 2458 CrossRef CAS.
-
(a) For selected reviews on organomagnesium catalyzed alkylating reactions, see: C. E. I. Knappke and V. W. A. Jacobi, Chem. Soc. Rev., 2011, 40, 4948 RSC;
(b) J. Terao and N. Kambe, Acc. Chem. Res., 2008, 41, 1545 CrossRef CAS;
(c) A. F. Pozharskii and O. V. Ryabtsova, Russ. Chem. Rev., 2006, 75, 709 CrossRef CAS;
(d) R. W. Hoffmann, Chem. Soc. Rev., 2003, 32, 225 RSC.
-
(a) M. Arrowsmith, M. R. Crimmin, A. G. M. Barrett, M. S. Hill, G. Kociok-Köhn and P. A. Procopiou, Organometallics, 2011, 30, 1493 CrossRef CAS;
(b) S. R. Neal, A. Ellern and A. D. Sadow, J. Organomet. Chem., 2011, 696, 228 CrossRef CAS;
(c) J. F. Dunne, D. B. Fulton, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2010, 132, 17680 CrossRef CAS;
(d) X. Zhang, T. J. Emge and K. C. Hultzsch, Organometallics, 2010, 29, 5871 CrossRef CAS;
(e) M. R. Crimmin, M. Arrowsmith, A. G. M. Barrett, I. J. Casely, M. S. Hill and P. A. Procopiou, J. Am. Chem. Soc., 2009, 131, 9670 CrossRef CAS;
(f) P. Horrillo-Martínez and K. C. Hultzsch, Tetrahedron Lett., 2009, 50, 2054 CrossRef;
(g) X. Zhang, T. J. Emge and K. C. Hultzsch, Angew. Chem., Int. Ed., 2012, 51, 394 CrossRef CAS.
-
(a) For selected examples on organomagnesium catalyzed polymerization reactions, see: Y. Sarazin, M. Schormann and M. Bochmann, Organometallics, 2004, 23, 3296 CrossRef CAS;
(b) L. F. Sanchez-Barba, A. Garces, M. Fajardo and C. Alonse-Moreno, Organometallics, 2007, 26, 6403 CrossRef CAS;
(c) A. Garces, L. F. Sanchez-Barba and C. Alonso-Moreno, Inorg. Chem., 2010, 49, 2859 CrossRef CAS;
(d) J. D. Monegan and S. D. Bunge, Inorg. Chem., 2009, 49, 3248 CrossRef.
-
(a) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229 CrossRef CAS;
(b) A. G. M. Barrett, I. J. Casely, M. R. Crimmin and M. S. Hill, Inorg. Chem., 2009, 48, 4445 CrossRef CAS;
(c) V. C. Gibosn, J. A. Segal, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2000, 122, 7120 CrossRef;
(d) S. P. Green, C. Jones and A. Stasch, Angew. Chem., Int. Ed., 2008, 47, 9079 CrossRef CAS.
-
(a) M. H. Chisholm, J. Galluccl and K. Phomphral, Inorg. Chem., 2002, 41, 2785 CrossRef CAS;
(b) M. H. Chisholm, J. C. Gallucci and K. Phomphral, Inorg. Chem., 2004, 43, 275 CrossRef;
(c) W. C. Hung and C. C. Lin, Inorg. Chem., 2009, 48, 728 CrossRef CAS;
(d) M. H. Chisholm, J. C. Huffman and K. Phomphrai, J. Chem. Soc., Dalton Trans., 2001, 222 RSC.
-
(a) L. C. Liang, P. Y. Lee, W. L. Lan and C. H. Hung, J. Organomet. Chem., 2004, 689, 947 CrossRef CAS;
(b) H. Hao, S. Bhandari, Y. Ding, H. W. Roesky, J. Magull, H. G. Schmidt, M. Noltemeyer and C. Cui, Eur. J. Inorg. Chem., 2002, 1060 CrossRef CAS;
(c) J. Lewinski, M. Dranka, I. Kraszewska, W. Sliwinski and I. Justyniak, Chem. Commun., 2005, 4935 RSC.
- R. Li, B. Moubaraki, K. S. Murray and S. Brooker, Eur. J. Inorg. Chem., 2009, 2851 CrossRef CAS.
- R. Q. Li, B. Moubaraki, K. S. Murray and S. Brooker, Dalton Trans., 2008, 6014 RSC.
- B. C. Xu, T. Hu, J. Q. Wu, N. H. Hu and Y. S. Li, Dalton Trans., 2009, 8854 RSC.
- G. L. Regina, R. Silvertri, M. Artico, A. Lavecchia, E. Novellino, Q. Befani, P. Turini and E. Agostinelli, J. Med. Chem., 2007, 50, 922 CrossRef.
-
(a) A. D. Schofield, M. L. Barros, M. G. Cushion, A. D. Schwarz and P. Mountford, Dalton Trans., 2009, 85 RSC;
(b) L. Wang and H. Ma, Macromolecules, 2010, 43, 6535 CrossRef CAS;
(c) V. Poirier, T. Roisnel, J. F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820 RSC;
(d) M. L. Shueh, Y. S. Wang, B. H. Huang, C. Y. Kuo and C. C. Lin, Macromolecules, 2004, 37, 5155 CrossRef CAS;
(e) M. Y. Shen, Y. L. Peng, W. C. Hung and C. C. Lin, Dalton Trans., 2009, 9906 RSC.
- M. H. Chisholm, N. W. Eilerts, J. C. Huffman, S. S. Iyer, M. Pacold and K. Phomphrai, J. Am. Chem. Soc., 2000, 122, 11845 CrossRef CAS.
- A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 14 CrossRef.
-
G. M. Sheldrick, SHELXS 97, Program for the Solution of Crystal Structures, University of Gottingen, Germany, 1997 Search PubMed.
-
G. M. Sheldrick, SHELXL 97, Program for the Refinement of Crystal Structures, University of Gottingen, Germany, 1997 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.