Bicyclic ketone mediated synthesis of oxygenated aromatic systems†
Ramendra
Pratap
*a,
Resmi
Raghunandan
c,
Abhinav
Kumar
b and
Vishnu Ji
Ram
*b
aDepartment of Chemistry, University of Delhi, North Campus, Delhi 110007, India. E-mail: ramendrapratap@gmail.com
bDepartment of Chemistry, Lucknow University, Lucknow 226007, India. E-mail: vjiram@yahoo.com
cHASYLAB, Deutsches Elektronen-Synchrotron (DESY), A Research Centre of the Helmholtz Association, Notkestraße 85, D-22607, Hamburg
First published on 16th February 2012
Abstract
A concise and efficient synthesis of various oxygenated, polycyclic aromatic systems has been delineated through base catalyzed ring transformation of 2-oxo-4-(piperidin-1-yl)-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles by bicyclic ketones, as a source of carbanions in excellent yields.
The presence of heteroatoms in a polycyclic aromatic system increases the polarity, solubility and bioavailability1a of the compound. Polycyclic oxaheteroaromatics are also an environmental pollutant, formed and released into the atmosphere by various anthropogenic sources. Attention on the toxic effects of polycyclic oxaheterocycles has not been focused earlier because of their low concentration compared to polycyclic aromatic hydrocarbons.1b However, with growing awareness of the toxic effects of various environmental pollutants, attention has been focused on understanding the toxicity of polycyclic oxaheteroaromatics on human health and ecological balance2 together with their introduction to the environment, soil, sediments, and water. Like polycyclic aromatic hydrocarbons, oxaheterocycles may also be metabolized by cytochrome P450 to diol epoxides which covalently bind with the amino functions of purine bases of cellular DNA through C–N linkages3,4 and thereby alter DNA replication, resulting in carcinogenesis.
As evident from the topography of various designed polycyclic oxaheteroaromatics (I–III), they possess ‘bay’ as well as ‘fjord’ regions, and are important in activation ultimately to tumorigenic diol epoxide metabolites.4 These metabolites are highly potent mammalian cell mutagens.5 All the prototype structures I, II and III have a close resemblance with benzo[c]phenanthrene IV, which is a carcinogen, Fig. 1.
![Various designed oxaheteroaromatics I–III and benzo[c]phenanthrene (IV).](/image/article/2012/RA/c2ra01253g/c2ra01253g-f1.gif) | ||
| Fig. 1 Various designed oxaheteroaromatics I–III and benzo[c]phenanthrene (IV). | ||
Past observations on the tumorigenic properties of various polycyclic arenes and heteroarenes evidence that partial reduction reduces or destroys the mutagenic properties of the molecule by disrupting the coplanarity.6 These observations prompted us to synthesize partially reduced angular benzofuran based oxaheterocycles to reduce the toxicity of the compounds. However no toxicity data are reported.
Herein, we report a novel approach for the construction of partially reduced polycyclic oxaheterocycles through base catalyzed ring transformation of 2-oxo-4-(piperidin-1-yl)-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles 1 with 2,3-dihydrobenzofuran-3-one 5, 4,5,6,7-tetrahydrobenzo furan-4-one 8 and 1-benzosuberone 11 separately. The 2-oxo-4-(piperidin-1-yl)-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles used as precursors for the synthesis of various angular oxaheterocycles were prepared7 in two steps. The first step was the synthesis of 2-oxo-4-methylsulfanyl-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles by base catalyzed reaction of 1-tetralone and methyl 2-cyano-3,3-dimethylthioacrylate, which on amination with piperidine in boiling ethanol afforded 2-oxo-4-(piperidin-1-yl)-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles 1. The preference to use 1 as a precursor was to avoid the side reactions at C-4 of the 2-oxo-4-methylsulfanyl-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles as well as to obtain clean partially reduced ring transformed products. Our synthetic strategy was to start with dihydro precursors 1 to obtain partially reduced various angular polycyclic oxaheterocycles, as selective reduction at the final stage is very difficult and may lead to a complex mixture of reduced products.
As is evident from the topography of 2-oxo-4-(piperidin-1-yl)-5,6-dihydro-2H-benzo[h]chromene-3-carbonitriles 1, they possess three electrophilic centres C-2, C-4 and C-10b, in which latter is highly electrophilic in nature due to extended conjugation and the presence of an electron-withdrawing CN substituent at position 3 of the chromene ring and consequently prone to nucleophilic attack. The nucleophiles used were carbanions generated in situ from cyclohexanone and various bicyclic ketones such as 2,3-dihydrobenzofuran-3-one 5, 4,5,6,7-tetrahydrobenzofuran-4-one 8 and benzosuberone 11 to observe the impact of monocyclic and bicyclic ketones on the course of the reaction. Thus, stirring an equimolar mixture of 1, cyclohexanone 2 and powdered KOH in DMF at room temperature for 2–3 h, followed by usual work-up and purification of the crude product by column chromatography, afforded 6-piperidin-1-yl-1,2,3,4,7,8-hexahydrobenzo[c]phenanthrene-5-carbonitrile 3 in excellent yield without any formation of product 4 (Scheme 1).8 However, the reaction of 1 with 2,3-dihydrobenzofuran-3-one 5 exclusively gave 5,6-dihydro-8,13-dioxaindeno[1,2-c]phenanthren-7-ylidene)acetonitrile 6.while reaction of 1 with 1-indanone provided only indeno[1,2-c] phenanthrene8c7 without any formation of product like 6 (Scheme 2). These reactions demonstrate the pivotal role of a ring oxygen in the stabilization of the enolate through conjugation, responsible for the formation of products like 6. From these experiments, it is concluded that monocyclic ketones are not suitable precursors for the synthesis of oxaheteroaromatics, so we used only bicyclic ketones for our further studies.
![Synthesis of 6-sec-amino-1,2,3,4,7,8-hexahydrobenzo[c]phenanthrene-5-carbonitriles 3.](/image/article/2012/RA/c2ra01253g/c2ra01253g-s1.gif) | ||
| Scheme 1 Synthesis of 6-sec-amino-1,2,3,4,7,8-hexahydrobenzo[c]phenanthrene-5-carbonitriles 3. | ||
![Synthesis and yield optimization of (5,6-dihydro-8,13-dioxaindeno[1,2-c]phenanthren-7-ylidene)acetonitriles 6.](/image/article/2012/RA/c2ra01253g/c2ra01253g-s2.gif) | ||
| Scheme 2 Synthesis and yield optimization of (5,6-dihydro-8,13-dioxaindeno[1,2-c]phenanthren-7-ylidene)acetonitriles 6. | ||
Similarly the ring transformation of 1 by 4,5,6,7-tetrahydrobenzofuran-4-one 8 provided pentacyclic (6,7-dihydro-1,4-dioxabenzo[g]cyclopenta[a]phenanthren-5-ylidene)acetonitrile 9 in moderate yield as a major product. The minor product isolated was also characterized as 6,7,12,13-tetrahydro-5-(piperidin-1-yl)-1-oxacyclopenta[a]benzo[c]-phenanthrene-4-carbonitrile 10 in 18–21% yields. The formation of products 9 and 10 from this experiment is only possible if the reaction follows both the paths A and B as shown in Scheme 3.
![Proposed mechanism involved in the synthesis of (6,7-dihydro-1,4-dioxabenzo[g]cyclopenta[a]phenanthren-5-ylidene)acetonitrile 9 and 6,7,12,13-tetrahydro-5-(piperidin-1-yl)-1-oxacyclopenta[a]benzo[c]-phenanthrene-4-carbonitrile 10.](/image/article/2012/RA/c2ra01253g/c2ra01253g-s3.gif) | ||
| Scheme 3 Proposed mechanism involved in the synthesis of (6,7-dihydro-1,4-dioxabenzo[g]cyclopenta[a]phenanthren-5-ylidene)acetonitrile 9 and 6,7,12,13-tetrahydro-5-(piperidin-1-yl)-1-oxacyclopenta[a]benzo[c]-phenanthrene-4-carbonitrile 10. | ||
The initial step in the formation of both the products 9 and 10 is the Michael addition of cyclic ketone 8 to 1 and thereafter, it may follow either path A or B. In the formation of major product 9, reaction proceeds possibly through intramolecular enolate addition to enamine followed by loss of carbon dioxide and piperidine. The most crucial step in the formation of 9 is oxidation of C–C bond 12–13. In the formation of 10 there is an intramolecular enamine addition to the carbonyl function with loss of water as shown in Scheme 3. From this reaction it was concluded that piperidine acts as a better leaving group and formation of oxaheteroaromatic 9 is favored over 10. It is conspicuous that during cyclization, the dihydrobenzofuran ring was aromatized in the case of 9, while it remained unaffected for 10. Based on this result, we proposed that the driving force for oxidation is the presence of the electron-withdrawing cyano group in extended conjugation, which facilitates the aromatization.
The generality of the reaction was further explored, using benzosuberone 11 as a carbanion source for the ring transformation of 1 (Scheme 4). Thus, reactions of 1 with benzosuberone 11 under analogous reaction conditions gave only (5,6-dihydro-14H-8-oxabenzosuberano[1,2-c]phenanthren-7-ylidene)acetonitriles 12 in good yields, possibly due to the stability of the intermediate formed by attack of the carbanion at C-10b. It seems that the keto intermediate in this case is less stable compared to the enolate, which is responsible for the formation of an usual ring transformed product. Representative compounds are characterized by spectroscopic analysis.9
![Synthesis of (5,6-dihydro-14H-8-oxabenzosuberano[1,2-c]phenanthren-7-ylidene)acetonitriles 12.](/image/article/2012/RA/c2ra01253g/c2ra01253g-s4.gif) | ||
| Scheme 4 Synthesis of (5,6-dihydro-14H-8-oxabenzosuberano[1,2-c]phenanthren-7-ylidene)acetonitriles 12. | ||
X-Ray diffraction studies10 of 9a revealed that the compound is nonplanar and acquires helical conformation. The conformation of the title compound with arbitrary numbering is shown as an ORTEP diagram (Fig. 2). Crystal packing of 9a reveals the presence of four molecules in the unit cell in the form of enantiomeric pairs, in which one of the isomers has a tendency to acquire a clockwise helical conformation while the other is anticlockwise, as evident from Fig. 2.
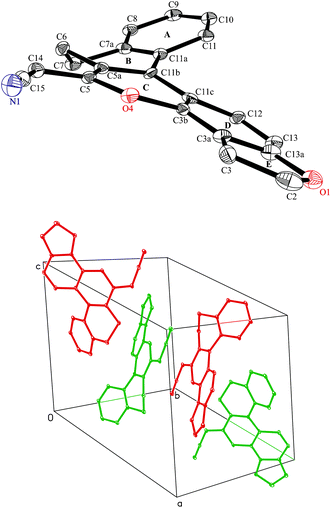 | ||
| Fig. 2 Displacement ellipsoid plot (30% probability) showing the molecular structure of 9a with atomic labelling and arrangement of molecules in the unit cell. | ||
The torsion angles (C11–C11a–C11b–C11c and C12–C11c–C11b–C11a) of compound 9a are −31.31 and −16.69°. The inner C–C bond length has increased while the outer has remained closer to normal single and double bond lengths due to stretching and bending of the chemical bonds, as shown in Table 1. The presence of a 5-membered fused ring at the C3–C13a position enhances the degree of non-planarity in the compound by ∼48°.
| Inner C–C bond lengths | Outer C–C bond lengths |
|---|---|
| C11–C11a 1.446 | O4–C5 1.354 |
| C11a–C11b 1.433 | C5–C5a 1.458 |
| C11b–C11c 1.481 | C5a–C6 1.458 |
| C11c–C12 1.443 | C6–C7 1.563 |
In summary, this protocol provides an easy access to the synthesis of various mono and dioxa polycyclic aromatic systems through the base catalyzed ring transformation of suitably functionalized 2-oxobenzo[h]chromenes by bicyclic ketones in moderate to good yields. The synthetic strategy provides a simple route for the construction of various mono and dioxa polycyclic aromatic systems without use of expensive catalysts or reagents. Moreover, work-up of the reaction is very simple and convenient. A single crystal X-ray diffraction study for 9a also shows that partial reduction in polycyclic ring systems increases the degree of helicity and could be useful to reduce the carcinogenicity. These molecules are still under investigation for their carcinogenic properties.
Acknowledgements
The authors are thankful to CSIR, New Delhi and UGC, New Delhi for financial support. RP is thankful to the University of Delhi, India for providing an R and D grant.References
- (a) J. D. Cooney and C. W. Gehrs, Aquat. Toxicol., 1984, 5, 197–209 Search PubMed; (b) T. K. Nielson, C. H. Sigur, O. Jorgensen, P. Hansen and U. Kirso, Environ. Sci. Technol., 1997, 31, 1102–1108 CrossRef CAS.
- E. A. J. Bleeker, S. Wiegman, P. de Voogt, M. Kraak, H. A. Leslie, H. de Hass and W. Rev Admiraal, Environ. Contam. Toxicol., 2002, 173, 39–84 Search PubMed.
- (a) M. F. Denisenko, A. Pao, M. Tang and G. P. Pfeifer, Science, 1996, 274, 430–432 CrossRef CAS; (b) G. P. Pfeifer and M. F. Denissenko, Environ. Mol. Carcin., 1998, 31, 197–205 Search PubMed; (c) M. F. Denissenko, J. X. Cheng, M. Tang and G. P. Pfeifer, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 3893–3898 CrossRef CAS; (d) J. X. Cheng, Y. Zheng, M. West and M. Tang, Cancer Res., 1998, 58, 2170–2175 Search PubMed.
- (a) C. C. Cheng, Structural aspects of antineoplastic agents-a new approach, in Progress in Medicinal Chemistry, ed. G. P. Ellis and G. B. West, Elsevier Science Publisher, B. V. (Biomedical Division), Amsterdam, 1988, vol. 25, pp. 35–83 Search PubMed; (b) T. Buterin, M. T. Hess, N. Luneva, N. E. Geacintov, S. Amin, H. Kroth, A. Seidel and H. Naegeli, Cancer Res., 2000, 60, 1849–1856 Search PubMed; (c) S. Amin, D. Desai and S. S. Hecht, Carcinogenesis, 1993, 14, 2033–2037 Search PubMed.
- E. R. Butch, H. Yagi, D. M. Jarina, S. Amin and W. M. Baird, Polycyclic Aromat. Compd., 1994, 6, 63–70 Search PubMed.
- D. D. Phillips and A. W. Johnson, J. Am. Chem. Soc., 1955, 77, 5477–5983 Search PubMed.
- (a) R. Pratap and V. J. Ram, Tetrahedron Lett., 2007, 48, 1715–1719 Search PubMed; (b) R. Pratap and V. J. Ram, Tetrahedron, 2007, 63, 10300–10308 Search PubMed; (c) R. Pratap and V. J. Ram, Tetrahedron Lett., 2009, 50, 2805–2807 Search PubMed.
- (a) R. Pratap and V. J. Ram, Tetrahedron Lett., 2009, 50, 4239–4242 Search PubMed; (b) R. Pratap and V. J. Ram, Tetrahedron Lett., 2007, 48, 4379–4382 Search PubMed; (c) R. Pratap, R. Raghunandan, P. R. Maulik and V. J. Ram, Tetrahedron, 2010, 66, 1458–1464 Search PubMed.
- General procedure for the synthesis of (5,6-dihydro-8,13-dioxaindeno[1,2-c]phenanthren-7-ylidene)acetonitriles 6: a mixture of 1 (0.5 mmol), 2,3-dihydrobenzofuran-3-one 5 (0.6 mmol) and powdered KOH (0.8 mmol) in DMF was stirred at room temperature for 2 h. During this period all the starting material was consumed with the appearance of a new spot on the TLC. Thereafter, the reaction mixture was poured onto crushed ice with vigorous stirring, followed by neutralization with 10% HCl. The resulting precipitate was filtered, washed with water, dried and purified through a neutral alumina column using 2% ethyl acetate in hexane as eluent. (6a). Yield 72%; mp 182–184 °C; Rf 0.65 (CHCl3); 1H NMR (CDCl3, 300 MHz): δ 2.49 (t, J = 7.4 Hz, 2H, CH2), 2.96 (t, J = 7.8 Hz, 2H, CH2), 4.57 (s, 1H, CH), 7.30–7.34 (m, 1H, ArH), 7.36–7.46 (m, 4H, ArH), 7.56 (d, J = 8.1 Hz, 1H, ArH), 7.80–7.83 (m, 1H, ArH), 8.34–8.41 (m, 1H, ArH); IR (KBr): 2198 cm−1; mass (ESI-MS) m/z 381.48 [M+ + 1]; anal calcd for C21H13NO2 272.33, C, 81.01; H, 4.21; N, 4.50; found C, 81.32; H, 4.11; N, 4.35%. General procedure for the synthesis of (6,7-dihydro-1,4-dioxabenzo[g]cyclopenta[a]phenanthren-5-ylidene)acetonitriles 9 and 6,7,12,13-tetrahydro-5-(piperidin-1-yl)-1-oxacyclopenta[a]benzo[c]phenanthren-4-carbonitriles 10: these were obtained by stirring a mixture of 1 (0.5 mmol), 4,5,6,7-tetrahydrobenzofuran-4-one 8 (0.6 mmol) and powdered KOH (0.8 mmol) in DMF at room temperature for 2–3 h. The reaction was monitored by TLC, which showed two spots, one with a high Rf and the other with a low Rf value. After completion of reaction, excess DMF was removed under reduced pressure. Thereafter, the reaction mixture was poured onto crushed ice with vigorous stirring, followed by neutralization with 10% HCl. The resulting precipitate was filtered, washed with water, dried and purified through a neutral alumina column. The compound with high Rf was eluted with 1.5% ethyl acetate in hexane and characterized as 10, while the compound with low Rf was eluted with 4% ethyl acetate in hexane and characterized as 9. (9a). Yield 61%; mp 202–204 °C; Rf 0.60 (CHCl3); 1H NMR (CDCl3, 300 MHz): δ 2.40 (t, J = 7.2 Hz, 2H, CH2), 2.85 (t, J = 7.2 Hz, 2H, CH2), 4.65 (s, 1H, CH), 7.17–7.20 (m, 1H, ArH), 7.32–7.41 (m, 4H, ArH), 7.65–7.70 (m, 1H, ArH), 7.74–7.80 (m, 2H, ArH); IR (KBr): 2201 cm−1; mass (ESI-MS) m/z 312 [M+ + 1]; anal calcd for C21H13NO2 272.33, C, 81.01; H, 4.21; N, 4.50; found C, 80.69; H, 4.01; N, 4.55%; (10a). Yield 21%; mp 180–182 °C; Rf 0.85 (CHCl3); 1H NMR (CDCl3, 300 MHz) δ 1.56–1.58 (m, 6H, CH2), 1.63–1.66 (m, 2H, CH2), 1.73–1.75 (m, 2H, CH2), 2.73–2.80 (m, 4H, CH2), 3.22–3.33 (m, 4H, CH2), 7.27–7.40 (m, 3H, ArH), 7.49–7.51 (m, 2H, ArH), 7.72–7.75 (m, 1H, ArH); IR (KBr): 2221 cm−1; mass (ESI-MS) m/z 312 [M+ + 1]; anal calcd for C26H24N2O 380.48, C, 82.07; H, 6.36; N, 7.36; found C, 81.97; H, 6.43; N, 7.40%. General procedure for the synthesis of (5,6-dihydro-14H-8-oxabenzosuberano[1,2-c]phenanthren-7-ylidene)acetonitrile 12: it was obtained by following the procedure described for the preparation of 6. The compound 12 was purified by neutral alumina column chromatography, using 30% chloroform in hexane as eluent. (12a). Yield 77%; mp 202–204 °C; Rf 0.85 (CHCl3); 1H NMR (CDCl3, 300 MHz): δ 2.39 (t, J = 7.23 Hz, 2H, CH2), 2.85 (t, J = 7.0 Hz, 2H, CH2), 3.20 (d, J = 7.9 Hz, 2H, CH2), 4.53 (s, 1H, CH), 5.84–5.92 (m, 1H, CH), 6.14 (d, J = 10.1 Hz, 1H, CH), 7.23–7.30 (m, 4H, ArH), 7.39–7.51 (m, 2H, ArH), 7.72–7.75 (m, 1H, ArH), 8.13 (dd, J = 1.8 and 1.4 Hz, 1H, ArH); 13C NMR (CDCl3, 75 MHz): δ 21.4, 26.4, 32.7, 63.1, 112.7, 117.3, 123.3, 123.5, 123.8, 125.0, 125.4, 125.7, 126.5, 126.6, 127.0, 127.9, 129.1, 129.8, 129.9, 136.9, 137.2, 138.4, 163.6; IR (KBr): 2191 cm−1; mass (ESI-MS) m/z 337 [M+ + 1]; anal calcd for C24H17NO 335.39, C, 85.94; H, 5.11; N, 4.18; found C, 86.11; H, 5.03; N, 4.19%.
- The crystal data of 9a: C20H10NO2, M = 296.29, monoclinic, P21/c, a = 12.388(2) Å, b = 12.6450(1) Å, c = 10.535(2) Å, β = 112.90(2)°, V = 1520.2(4) Å3, Z = 4, Dc = 1.295 g cm−3, μ(Mo-Kα) = 0.084 mm−1, F(000) = 612, rectangular block, yellow, 3427 reflections measured (Rint = 0.0533), 2666 unique, wR2 = 0.5108 for all data, conventional R = 0.1374 [(Δ/σ)max = 000)] on F-values of 1042 reflections with I > 2σ(I), S = 1.530 for all data and 218 parameters. Unit cell determination and intensity data collection (2θ = 50°) were performed on a Bruker P4 diffractometer at 293(2) K. Structure solutions by direct methods and refinements by full-matrix least-squares methods on F2. Programs: XSCANS [Siemens Analytical X-ray Instrument Inc., Madison, Wisconsin, USA, 1996], SHELXTL-NT [Bruker AXS Inc., Madison, Wisconsin, USA, 1997]. CCDC 838368.
Footnote |
| † CCDC reference number 838368. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra01253g |
| This journal is © The Royal Society of Chemistry 2012 |