Mononuclear and dinuclear complexes of manganese(III) and Iron(III) supported by 2-salicyloylhydrazono-1,3-dithiane ligand: synthesis, characterization and magnetic properties†
Weiwei
Zuo
,
Vitor
Rosa
,
Clarisse
Tourbillon
,
David
Specklin
,
Cheaib
Khaled
,
Mohamedally
Kurmoo
and
Richard
Welter
*
Laboratoire DECOMET, Institut de Chimie de Strasbourg, UMR 7177 CNRS, Université de Strasbourg, 4, rue Blaise Pascal, 67070, Strasbourg Cedex, France. Fax: +33 90 24 12 29; Tel: +33 90 24 15 93
First published on 7th February 2012
Abstract
The coordination chemistry of 2-salicyloylhydrazono-1,3-dithiane (H2L) was studied with manganese and iron ions. The following complexes have been isolated as crystalline materials, and their crystal structures have been determined by single crystal X-Ray crystallography: MnIII(acac)(HL)2 (1) (acac = acetylacetonate), MnIII(HL)3·CH2Cl2 (2), FeIII(HL)3·2CHCl3 (3), FeII(H2L)2Cl2·2CH3OH (4), FeIII2(μ-OMe)2(HL)4·0.5CH3OH (5), FeIII2(μ-O) (HL)4·3CH2Cl2 (6). All attempts to synthesize the dinuclear μ-methoxo complex [MnIII2(μ-OMe)2(HL)4] have so far failed, even when the procedure used in the case of 2-salicyloylhydrazono-1,3-dithiolane (H2L′) ligand, which worked very efficiently, was employed. The new iron dinuclear μ-methoxo complex (5) presented in this study shows antiferromagnetic intramolecular coupling (J = −21.1 cm−1), which is in agreement with the theoretical study proposed previously for its manganese analogue.
Introduction
The control of magnetic coupling in addition to the possibility of inducing a spin-state modification at the molecular scale is among the most challenging themes in the field of molecular magnetism. Considering a variety of magnetically interesting molecules,1 special attention has been paid to exchange coupled multinuclear paramagnetic complexes.2,3 Systems presenting lower nuclearity are of interest as, among others, versatile building blocks for cooperatively associated magnetic systems.4 For instance they can be incorporated in crystal lattices along with conducting molecules, which may lead to the formation of innovative magnetic/conducting bifunctional materials.5 Due to the dependence of coupling pathways on the structure and symmetry of the organic ligands, dinuclear molecular systems offer interesting possibilities to tune metal-to-metal interactions by subtle structural changes within the organic periphery. We have reported an interesting dinuclear MnIII complex MnIII2 (μ-OMe)2(HL′)4, which was found to exhibit the largest J value (J = + 19.7 cm−1) reported so far for a MnIII–MnIII interaction. The peculiarity of the MnIII2(μ-OMe)2(HL′)4 solid-state structure arises from an unsymmetrical arrangement of the ligands leading to orthogonality of the magnetic orbitals and consequently the ferromagnetic exchange coupling. This situation is certainly related to intramolecular non-classical H-bonds occurring between hydrogen atoms of the dithiolane rings and the centroids of the phenol groups.6 In addition to the MnIII chemistry, we have also studied the coordination chemistry of the 2-salicyloylhydrazono-1,3-dithiolane ligand to other metal ions such as iron,7,8cobalt9 and chromium10 to form a series of mononuclear and dinuclear complexes (Scheme 1). These complexes have shown interesting chemical and physical properties. For example, an unusual spontaneous reduction from FeIII to FeII of the mononuclear complexes was observed.7 This redox reaction was subsequently fully investigated by electrochemistry, EPR studies, magnetic measurements and solid-state molecular structure determination. Such a phenomenon opens up new research opportunities, as recently been pointed out by Wernsdorfer et al.11 and forms the basis of a patent.12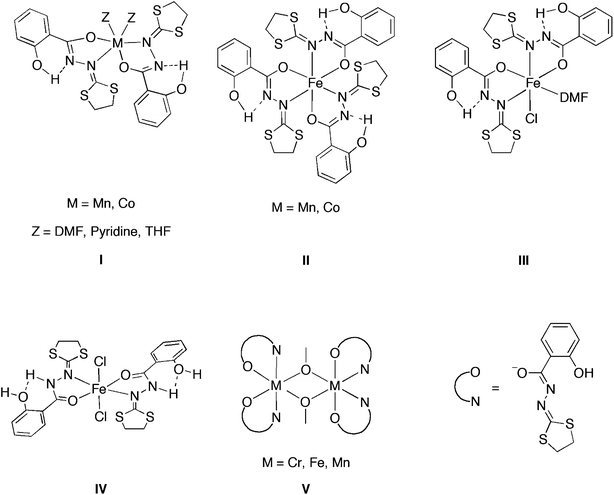 | ||
| Scheme 1 Transition metal complexes of 2-salicyloylhydrazono-1,3-dithiolane ligand. | ||
The asymmetric FeIII2(μ-OMe)2(HL′)4 complex has also been synthesized and characterized.7 This dinuclear FeIII compound shows antiferromagnetic intramolecular coupling, which is in agreement with the previously proposed theoretical model for the related MnIII complex.6 As predicted by DFT calculations, the CrIII μ-methoxo dinuclear complex featuring the same ligand also exhibited a strong antiferromagnetic coupling.10
Via minor modifications of the chelating ligand, it is expected to control the structural geometry of the complexes obtained, as well as their physical properties. We have thus started to explore the coordination chemistry of 2-salicyloylhydrazono-1,3-dithiane (H2L), which is closely related to H2L′ where it has a 1,3-dithiane group in place of the 1,3-dithiolane moiety.9,10 A dinuclear μ-methoxo CrIII2(μ-OMe)2(HL)4 complex, chelated by HL exhibiting a strong antiferromagnetic coupling, was successfully synthesized by the reaction of CrCl3 with this ligand in the presence of zinc.10 In addition, one novel CoII mononuclear complex and one diamagnetic μ-hydroxo dinuclear CoIII complex [CoIII2(μ-OH)2(HL)4] supported by the ligand HL were also stabilized and characterized.9
The hypothesis that the similarity between the structures of H2L and H2L′ would allow HL to be a suitable ligand to stabilize a dinuclear μ-methoxo MnIII complex, which is expected to exhibit very strong intramolecular ferromagnetic interaction, prompted us to extend the coordination chemistry of ligand H2L to MnIII. In this article, we report our attempts to synthesize the desired dinuclear μ-methoxo MnIII complex stabilized by 2-salicyloylhydrazono-1,3-dithiane. In addition, following a systematic approach and due to their similar coordination behaviour, we also describe the synthesis and characterization of four iron mononuclear and dinuclear complexes supported by HL and H2L, and the synthesis and magnetic property measurement of μ-methoxo dinuclear iron complex.
Results and discussion
Synthesis and characterization of manganese complexes MnIII(acac)(HL)2 (1) and MnIII(HL)3 (2)
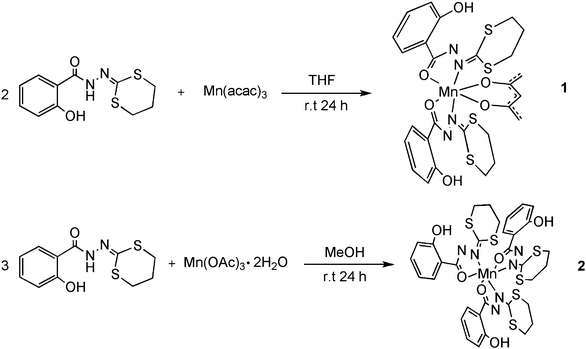 | ||
| Scheme 2 Synthesis of complexes MnIII(acac)(HL)2(1) and MnIII(HL)3 (2). | ||
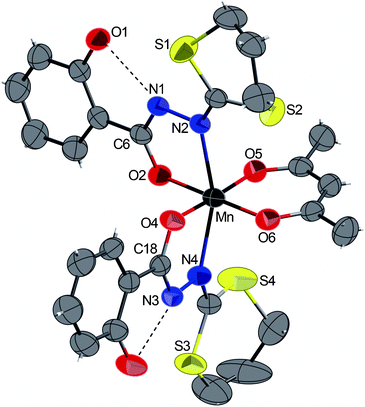 | ||
| Fig. 1 ORTEP view of the complex 1 with partial labelling scheme. The ellipsoids enclose 50% of the electronic density. Selected distances (Å) and angles (°): Mn–N2 2.261(2), Mn–N4 2.280(2), Mn–O2 1.922(2), Mn–O4 1.921(2), Mn–O5 1.912(3), Mn–O6 1.908(2); O5–Mn–O4 176.59(8), N2–Mn–N4 158.37(8), O6–Mn–O2 179.63(8). Dashed lines indicate intramolecular hydrogen bonds. | ||
The complex MnIII(HL)3·CH2Cl2 (2) was obtained in 31% yield by reacting three equivalents of H2L with manganese acetate dihydrate in methanol. Good quality crystals (but very sensitive to desolvation) were obtained by slow diffusion of pentane to a solution of the complex in dichloromethane (Scheme 1). In contrast to the case of 2-salicyloylhydrazono-1,3-dithiolane,6 and in spite of addition of excess base (NaOAc, NEt3etc.) we were unable to generate the μ-methoxo dinuclear complex MnIII2(μ-OMe)2(HL)4.
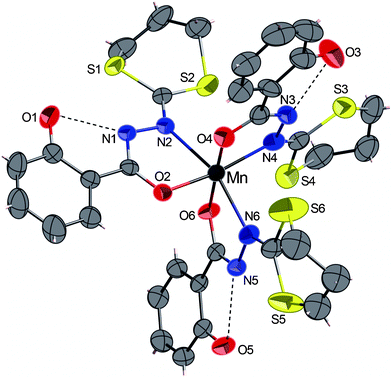 | ||
| Fig. 2 ORTEP view of the complex 2·CH2Cl2 with partial labelling scheme. The ellipsoids enclose 50% of the electronic density. The solvent CH2Cl2 was omitted for clarity. Selected distances (Å) and angles (°): Mn–N2 2.294(3), Mn–N4 2.088(4), Mn–N6 2.259(3), Mn–O2 1.922(3), Mn–O4 1.892(3), Mn–O6 1.894(3); O4–Mn–O6 177.3(2), O2–Mn–N4 167.3(1), N2–Mn–N6 161.5(1). | ||
The dark red complex 2·CH2Cl2 crystallizes in the triclinic centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The crystal structure (Fig. 2) shows that the MnIII ion is surrounded by three anionic bidentate HL− ligands, forming a distorted octahedral geometry of the metal centre. All distances are in agreement and very similar, to those measured in complex 1 and with other manganese complexes containing 2-salicyloylhydrazono-1,3-dithiolane ligand.13 As already observed in complex 1, the intramolecular O–H⋯N hydrogen bonds are established between OH groups of the phenol rings and the hydrazine groups. Besides the previously reported manganese complex supported by three 2-salicyloylhydrazono-1,3-dithiolane ligands,6 another very similar structure was observed in MnIII complex bearing three N′-benzylidenesalicylhydrazide ligands.16
. The crystal structure (Fig. 2) shows that the MnIII ion is surrounded by three anionic bidentate HL− ligands, forming a distorted octahedral geometry of the metal centre. All distances are in agreement and very similar, to those measured in complex 1 and with other manganese complexes containing 2-salicyloylhydrazono-1,3-dithiolane ligand.13 As already observed in complex 1, the intramolecular O–H⋯N hydrogen bonds are established between OH groups of the phenol rings and the hydrazine groups. Besides the previously reported manganese complex supported by three 2-salicyloylhydrazono-1,3-dithiolane ligands,6 another very similar structure was observed in MnIII complex bearing three N′-benzylidenesalicylhydrazide ligands.16
| Solvents | Non-solvents |
|---|---|
| CHCl3 | MeOH |
| CH2Cl2 | EtOH |
| ClCH2CH2Cl | i PrOH |
| DMF | Diethyl ether |
| THF | H2O |
The second attempt to synthesize the desired dinuclear complex was based on the assumption that there were little differences between the structures of the complex MnIII2(μ-OMe)2(HL′)4 and the desired structure MnIII2(μ-OMe)2(HL)4. Therefore, some tests have been set up to force the crystalline growth of MnIII2(μ-OMe)2(HL)4 instead of the complex 2. To do this, diverse proportions of powder from the reaction between two equivalents of HL′ with manganese(III) acetate dihydrate were added to a chloroform solution of the powder obtained from the same reactions with HL, followed by slow diffusion of methanol into this mixed solution. However, unfortunately the nucleation of the complex MnIII2(μ-OMe)2(HL′)4 did not seem to encourage the growth of MnIII2(μ-OMe)2(HL)4.
Synthesis and characterization of iron complexes
Following a systematic procedure and taking in consideration some shared characteristics of iron and manganese, we synthesized in parallel, with the same ligand four iron complexes using commercial crystalline FeCl3 and Fe(acac)3 as metal sources.The aim was to examine the possibility to obtain well-defined molecular complexes of FeIII bearing a H2L ligand and to compare their respective reactivity towards the same ligand.
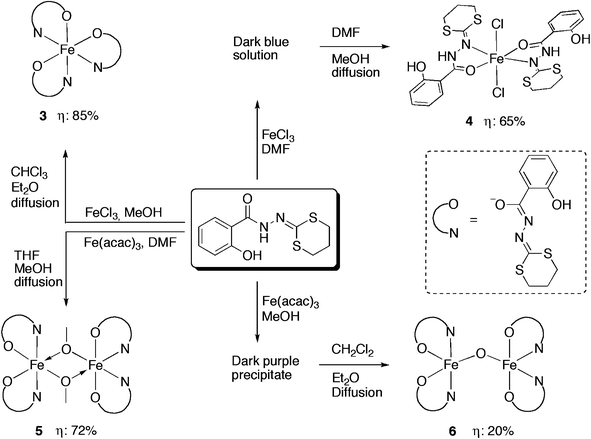 | ||
| Scheme 3 Synthetic methods for the iron complexes 3, 4, 5 and 6. | ||
Complex 3·2CHCl3 crystallizes in the triclinic space group P. The FeIII centre has a [N3O3] coordination sphere with a pseudo-octahedral geometry. The ligands HL− are coordinated to the iron as bidentate chelating agents via the nitrogen atoms (N2, N4, N6) and the carboxyl oxygen atoms (O2, O4, O6) of the hydrazones in a propeller configuration fashion. The arrangement of the 2-salicyloylhydrazono-1,3-dithiane ligands in this iron complex is similar to the analogous manganese complex 2. The Fe–N distances [2.123(3), 2.146(3) and 2.243(3) Å] and Fe–O distances [1.984(2), 1.957(2) and 1.952(2) Å] are similar to those found in the previously reported FeIII complex supported by 2-salicyloylhydrazono-1,3-dithiolane ligand.7 As shown in Fig. 3, the hydrogen bonding is donated from phenol O–H to a neighbouring nitrogen atom of the hydrazine group in each ligand. Several FeIII complexes containing an [N3O3] coordination sphere and similar geometrical features have been reported in the literature with o-iminobenzosemiquinonato,28o-aminophenol29 and 8-quinolinato.30
 | ||
| Fig. 3 ORTEP view of the complex 3·2CHCl3 with partial labelling scheme. The ellipsoids enclose 50% of the electronic density. The solvent molecules (CHCl3) were omitted for clarity. Selected distances (Å) and angles (°): Fe–N2 2.123(3), Fe–N4 2.146(3), Fe–N6 2.243(3), Fe–O2 1.984(2), Fe–O4 1.957(2), Fe–O6 1.952(2); O6–Fe–O4 165.7(1), N2–Fe–N4 158.5(2), O2–Fe–N6 165.3(1), O2–Fe–N2 76.7(1), O6–Fe–N6 74.7(1), O4–Fe–N4 76.6(1). | ||
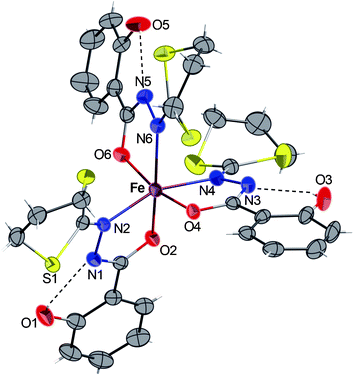
Complex 4·2CH3OH crystallizes in the monoclinic centrosymmetric space groupP 21/c. As shown in Fig. 4, the compound consists of a mononuclear complex of FeII. The presence of hydrogen atoms on the N1 and N1′ nitrogen atoms (clearly visible by Fourier differences and by the special conformation of the hydroxyl groups) indicates that both bidentate ligands are neutral. Considering both chloride atoms (−I oxidation state) complete the octahedral geometry, we consider that the iron cation, exhibit a +II oxidation state. In 4, the two bidendate H2L ligands are in a trans configuration, and the overall structure appears to be very similar to its analogues bearing benzoic hydrazide derivative ligands recently reported.7,31 In addition, the intramolecular H-bonds in each ligand (dashed lines in Fig. 4) were also detected and they are probably responsible for the quasi-planar orientation of the ligands. Complex 4 constitutes then a new example of iron(II) compound obtained via spontaneous reduction, which was already observed with the ligand H2L′ and related ligands as described in ref. 7 and 31.
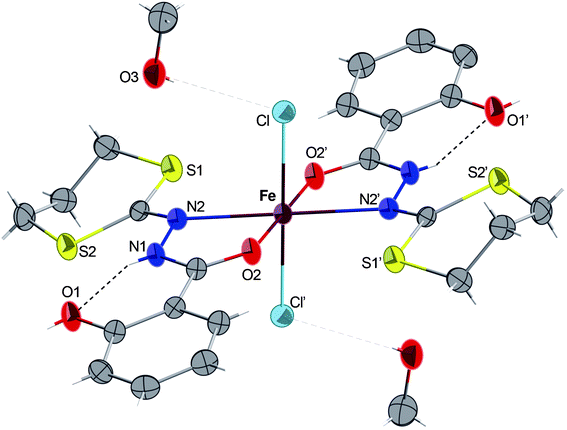 | ||
| Fig. 4 ORTEP view of the complex 4·2CH3OH with partial labelling scheme. The ellipsoids enclose 50% of the electronic density. Selected distances (Å) and angles (°): Fe–N2 2.292(2), Fe–O2 2.134(1), Fe–Cl 2.3995(5); O2–Fe–N2 73.15(4), Cl–Fe–Cl′ 180, O2–Fe–O2′ 180. Symmetry operator for equivalent positions: ′ = −x + 2, −y, −z + 2. | ||
Single crystals of complex 5·1/2CH3OH suitable for X-ray diffraction analysis were obtained by slow diffusion of methanol into a THF solution of the solid. Complex 5·1/2CH3OH crystallize in the orthorhombic space groupA b a 2. Exhibiting very similar structural features as its FeIII,7CrIII,10 and MnIII6 analogues, complexes 5 crystallizes as a neutral asymmetric dinuclear FeIII complex in which each iron metal centre is chelated by two HL− bidentate ligands and connected to each other via two μ-OMe groups (Fig. 5). The two metal ions Fe1 and Fe2 (which both lie on a crystallographic twofold axis) are in slightly distorted octahedral geometries but with different environments. Around Fe1, the oxygen atoms O2–O2′ are in a trans configuration and N2–N2′ in a cis configuration, while around Fe2, the oxygen atoms O4–O4′ are cis to one another and N4–N4′ in a trans configuration. As previously pointed out, this situation is most likely related to two intramolecular CH–π interactions arising from one 1,3-dithiane CH moiety and the centroid of the neighbouring phenol group, as indicated in Fig. 5. Finally, considering our systematic research on such dinuclear μ-OMe metal complexes, a complete magnetic study of 5 was carried out, and the results are described below.
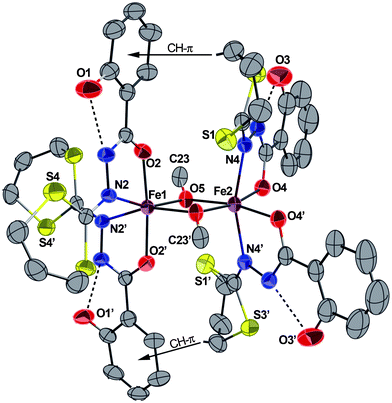 | ||
| Fig. 5 ORTEP view of the complex 5·1/2CH3OH with partial labelling scheme. H atoms and solvent molecules (CH3OH) are omitted for clarity. The ellipsoids enclose 50% of the electronic density. The dashed lines show representative intramolecular hydrogen bonds and the arrows represent the CH–π interactions. Selected distances (Å) and angles (°): Fe1–N2 2.196(3), Fe1–O2 1.972(2), Fe1–O5 1.975(2), Fe2–O5 1.994(2), Fe2–O4 1.977(2), Fe2–N4 2.154(3); O2–Fe1–O2′ 174.1(2), N2–Fe1–N2′ 95.2(2), N4–Fe2–N4′ 160.3(2), O4–Fe2–O4′ 98.7(2), Fe1–O5–Fe2 102.44(9). Symmetry operator for equivalent positions: ′ = −x + 1, −y, z. | ||
Complex 6·3CH2Cl2 crystallizes in the monoclinic space groupP 21/c (Fig. 6). The molecule contains a Fe–O–Fe core and the coordination geometry around each iron atom is described as distorted square pyramidal with the two hydrazine nitrogen atoms and the two carboxyl oxygen atoms of each ligand constituting the corners of the square plane and the μ-oxo oxygen atom occupying the axial position. No residual peak is detected in the Fourier difference map indicating unambiguously the absence of the hydrogen atom on the oxo oxygen atom. Also, the FeN2O2 coordination plane in each ironIII is oriented trans to the other relative to the oxo bridge, having a Fe–O–Fe bond angle of 167.1(2)°, which is in the normal range (135–180°) found in other closely related μ-oxo monobridged diiron complexes, where this angle is usually dominated by the steric parameters of the chelated ligands.19,32–35 The Fe⋯Fe distance in 6 (3.475 Å) is in the same range as those already reported for complexes with Fe–O–Fe cores (3.39–3.56 Å).17,24,33,36 The Fe–(μ-O) bond lengths observed [Fe1–O9 1.747(3), Fe2–O9 1.750(3) Å] are in the range found for other (μ-oxo)diironIII complexes (1.75–1.80 Å)17,20,33,37–39 but significantly shorter than that found in Fe–(μ-CH3O) of complex 5 (1.975(2) Å). Similar to complex 5, each iron atom is bound to two bidendate ligands in their basic forms and the geometry of the hydrazine chelating is comparable to those observed in either the mononuclear 3 or dinuclear μ-methoxo complex 5. Both the Fe–O(carboxyl) and Fe–N(hydrazine) bond distances are shorter than their counterparts in complex 5, probably due to the lower steric pressures at the metal centres in complex 6 than those in 5. No specific intramolecular CH–π interaction has been detected and only classical H–bonds occur between the OH groups of the phenol rings and the hydrazine groups.
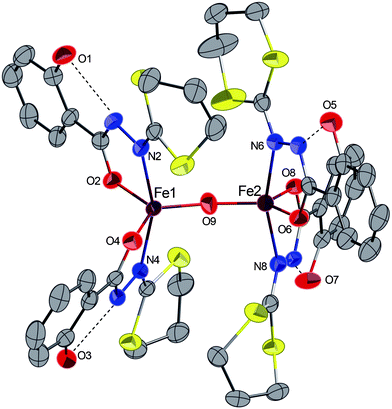 | ||
| Fig. 6 ORTEP view of the complex 6·3CH2Cl2 with partial labelling scheme. H atoms and solvent molecules (CH2Cl2) were omitted for clarity. The ellipsoids enclose 50% of the electronic density. The dashed lines show representative intramolecular hydrogen bonds and the arrows represent the CH–π interactions. Selected distances (Å) and angles (°): Fe1–N2 2.135(3), Fe1–O2 1.948(3), Fe1–N4 2.131(3), Fe1–O4 1.946(3), Fe1–O9 1.747(3), Fe2–O9 1.750(3), Fe2–N6 2.126(3), Fe2–O6 1.960(3), Fe2–N8 2.120(3), Fe2–O8 1.954(3); N4–Fe1–N2 157.6(2), O4–Fe1–O2 119.2(1), Fe1–O9–Fe2 167.1(2), N8–Fe2–N6 161.0(2), O8–Fe2–O6 119.8(2). | ||
| Compound reference | 1 | 2·CH2Cl2 | 3·2(CHCl3) | 4·2(CH3OH) | 5·1/2(CH3OH) | 6·3(CH2Cl2) |
|---|---|---|---|---|---|---|
| Chemical formula | C27H29MnN4O6S4 | C33H33MnN6O6S6·CH2Cl2 | C33H33FeN6O6S6·2(CHCl3) | C22H24Cl2FeN4O4S4·2(CH40) | C46H50Fe2N8O10S8·1/2(CH3O) | C44H44Fe2N8O9S8·3(CH2Cl2) |
| Formula Mass | 688.72 | 941.88 | 1096.60 | 727.53 | 1258.64 | 1451.83 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic | Orthorhombic | Monoclinic |
| a/[Å] | 11.712(5) | 10.429(2) | 11.227(3) | 10.394(4) | 23.038(2) | 12.8070(2) |
| b [Å] | 12.670(5) | 11.778(2) | 13.922(7) | 9.405(4) | 10.461(6) | 32.3360(8) |
| c [Å] | 20.965(5) | 17.263(3) | 15.914(6) | 17.332(3) | 22.989(6) | 15.2010(3) |
| α [°] | 90.00 | 105.39(5) | 69.021(2) | 90.00 | 90.00 | 90.00 |
| β [°] | 98.49(1) | 91.37(5) | 89.488(2) | 114.676(19) | 90.00 | 105.1750(11) |
| γ [°] | 90.00 | 90.35(5) | 77.920(2) | 90.00 | 90.00 | 90.00 |
| V [Å3] | 3076.9(19) | 2043.6(6) | 2265.1(15) | 1539.58(95) | 5540(4) | 6075.6(2) |
| Space group | P21/c |
P
|
P
|
P21/c | Aba2 | P21/c |
| Z | 4 | 2 | 2 | 2 | 4 | 4 |
| μ (Mo-Kα) [mm−1] | 0.748 | 0.812 | 1.014 | 0.980 | 0.888 | 1.075 |
| F(000) | 1424 | 968 | 1118 | 752 | 2602 | 2968 |
| Crystal size [mm] | 0.13 × 0.11 × 0.10 | 0.20 × 0.14 × 0.12 | 0.15 × 0.12 × 0.10 | 0.10 × 0.10 × 0.10 | 0.10 × 0.10 × 0.10 | 0.14 × 0.12 × 0.10 |
| θ min–θmax | 1.76–30.03 | 1.22–30.04 | 1.69–27.51 | 2.16–30.01 | 1.77–30.03 | 1.26–27.48 |
| Data set [h, k, l] | −15/16, −15/17, −23/29 | −14/14, −16/16, −21/24 | −14/14, −18/16, −20/20 | −12/14, −13/11, −24/24 | −32/31, −14/14, −29/32 | −16/11, −38/41, −19/19 |
| Total, unique data, Rint | 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 981, 8992, 0.0503 981, 8992, 0.0503 |
24068, 11916, 0.0375 |
19566, 10304, 0.0420 | 14463, 4496, 0.0773 |
25825, 7431, 0.0592 |
37050, 13388, 0.0656 |
| Observed data [I > 2σ(I)] | 5256 | 8810 | 7683 | 3713 | 5424 | 8532 |
| No. reflections, no. parameters | 8992, 379 | 11916, 490 |
10304, 541 |
4496, 191 | 7431, 350 | 13388, 721 |
| Final R1 values (I > 2σ(I)) | 0.0535 | 0.0847 | 0.0545 | 0.0395 | 0.0451 | 0.0576 |
| Final wR(F2) values (I > 2σ(I)) | 0.1308 | 0.1906 | 0.1156 | 0.1004 | 0.1106 | 0.1645 |
| Final R2 values (all data) | 0.1079 | 0.1139 | 0.0817 | 0.0504 | 0.0736 | 0.1034 |
| Final wR(F2) values (all data) | 0.1543 | 0.2064 | 0.1336 | 0.1070 | 0.1250 | 0.2001 |
| Goodness of fit on F2 | 1.007 | 1.068 | 1.048 | 1.070 | 1.007 | 1.082 |
| Max. and av. shift/error | 0.000/0.000 | 0.001/0.000 | 0.001/0.001 | 0.002/0.000 | 0.001/0.000 | 0.003/0.000 |
| Min, max residual density [e Å−3] | −0.711/0.603 | −0.692/2.169 | −1.611/1.787 | −0.916/0.573 | −0.532/0.588 | −2.018/1.971 |
Magnetic measurements of the μ-methoxo dinuclear iron complex 5
The magnetic property of complex 5 was investigated in the solid state in the 1.8–300 K temperature range and an applied field of 5 kOe (Fig. 7). The χT value at room temperature (6.62 emu K mol−1) is smaller than the expected value for two high spin FeIII (8.75 emu K mol−1 assuming g = 2). Upon cooling, the χT product increases to a maximum then decreases continuously to almost 0 (0.027 emu K mol−1 at 1.8 K). This indicates the occurrence of a relatively strong antiferromagnetic intramolecular interaction between the two FeIII ions. There is a small contribution of an impurity that gives the sharp rise at the lowest end of temperatures. The data were fit using the following spin Hamiltonian where all parameters have their usual meaning and the spin operator S is defined as S = SFe1 + SFe2:H = −JSFe1SFe2 + gβHS
To reproduce the data satisfactorily over the whole temperature region, including the small increase at the lowest temperatures, we consider a certain amount (ρ) of paramagnetic impurity (Simpure = 5/2). The fit leads the following values: J = −21.1(1) cm−1, g = 2.06(1) and ρ = 1.38(5)% with a good agreement factor R = 3.15 × 10−5 where R = (χexp − χcalc)2/χexp2.
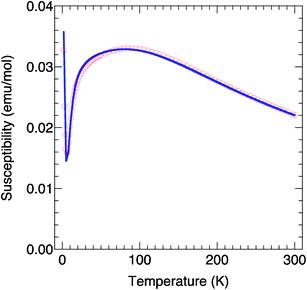 | ||
| Fig. 7 χ (circles) as a function of temperature for 5 and dark line corresponds to the fit of the χ = f(T) curve (see text). | ||
Conclusions
In this work, we have demonstrated that the reactions of 2-salicyloylhydrazono-1,3-dithiane (H2L) with different iron and manganese metal precursors under various conditions resulted in a series of new mononuclear or dinuclear complexes in good yields. All new complexes have been fully characterized by single crystal X-ray diffraction. The asymmetric dinuclear FeIII2(μ-OMe)2(HL)4 complex shows strong antiferromagnetic intramolecular coupling, which agrees with the previously proposed theoretical model for the related MnIII complex.6Following the previous findings that dinuclear μ-methoxo MnIII2(μ-OMe)2(HL′)4 presents one of the strongest intramolecular ferromagnetic interactions, our aim was the modulation of the magnetic interaction by small modifications of the peripheral ligand justifying the use of the 2-salicyloylhydrazono-1,3-dithiane ligand (H2L). The similarity between the structures of H2L and H2L′would allow H2L to be a suitable ligand to stabilize a dinuclear μ-methoxo MnIII complex, which is expected to also exhibit a very strong intramolecular ferromagnetic interaction. However, we failed in the synthesis of such desired complex although many attempts have been made. We are a bit puzzled as we have successfully synthesized and characterized the dinuclear μ-methoxo FeIII, MnIII and CrIII complexes chelated by HL′− and in addition, dinuclear FeIII and CrIII complexes supported by HL− were also successfully produced. Attempts are currently made in our laboratory, applying different synthetic methods and with the support of theoretical calculations, to achieve a better understanding of the different reactivity of the manganese when compared with other metals.
Experimental
General procedures
All manipulations were performed under aerobic conditions, using reagents and solvents as received. IR spectra were recorded in the region 4000–400 cm−1 on a Nicolet 6700 FT-IR spectrometer (ATR mode, diamond crystal). Elemental analysis was performed by the “Service de microanalyses”, Université de Strasbourg. The ligand 2-salicyloylhydrazono-1,3-dithiane was prepared according to the reported procedure.10Synthesis of MnIII(acac)(HL)2 (1)
To a solution of 2-salicyloylhydrazono-1,3-dithiane (0.20 g, 0.74 mmol) in THF (20 mL) was added a solution of manganese acetylacetonate (0.13 g, 0.36 mmol) under stirring. The dark solution was further stirred at room temperature for 24 h, then the solvent was removed by vacuum and the resulting solid was washed with methanol and dried (yield: 0.17 g, 67%). Dark red single crystals suitable for X-ray analysis were obtained by slow diffusion of methanol in a chloroform solution of the complex. IR (pure, orbit diamond, cm−1): 2361, 1517. Anal. Calcd. for C27H29MnN4O6S4: C, 47.08; H, 4.24; N, 8.13. Found: C, 46.78; H, 4.17; N 7.79.Synthesis of MnIII(HL)3·CH2Cl2 (2)
To a solution of manganese acetate dihydrate (0.10 g, 0.36 mmol) in methanol (20 mL) was added 2-salicyloylhydrazono-1,3-dithiane (0.29 g, 1.08 mmol). The dark solution was further stirred at room temperature for 24 h before the solvent was removed under vacuum to afford a yellow solid (yield: 0.10 g, 31%). Red rectangular crystals suitable for X-ray analysis were obtained by slow diffusion of methanol through a CH2Cl2 solution of the complex. IR (pure, orbit diamond, cm−1): 1588, 1515. Anal. Calcd. for C33H33MnN6O6S6·CH2Cl2: C, 43.35; H, 3.75; N, 8.92. Found: C, 43.06; H, 3.68; N, 8.81.Synthesis of FeIII(HL)3·2CHCl3 (3)
FeCl3 (0.013 g, 0.08 mmol) was added to the solution of H2L (0.067 g, 0.25 mmol) dissolved in methanol (20 ml), and then the mixture was further stirred at room temperature for 24 h. After reaction the solvent was removed under vacuum to obtain a green powder. Red rectangular crystals of 3·2CHCl3 were obtained by slow diffusion of diethyl ether into the CHCl3 solution of the complex. (Yield: 0.075 g, 85%). IR (pure, orbit diamond, cm−1): 1518, 1357, 1251, 754. Anal. Calc. for C33H33FeN6O6S6·2CHCl3: C, 38.33; H, 3.22; N, 7.66. Found: C, 38.51; H, 3.33; N 7.92.Synthesis of FeII(H2L)2Cl2·2CH3OH (4)
A yellow solution of FeCl3 (0.019 g, 0.12 mmol) in DMF (1 mL) was added to a well-stirred colourless solution of the ligand (0.064 g, 0.24 mmol) in DMF (2 mL). The colour changes immediately from yellow to dark blue. The homogeneous mixture was left under stirring overnight at room temperature. A yellow crystalline powder of the complex was obtained after a slow diffusion (3 to 4 days) of MeOH into the crude reaction mixture in a sealed glass tube (Yield: 0.052 g, 65%). Anal. Calc. for C22H24Cl2FeN4O4S4·2CH3OH: C, 39.62; H, 4.43; N, 7.70. Found: C, 39.71; H, 4,04; N 7.67.Synthesis of FeIII2(μ-OMe)2(HL)4·1/2CH3OH (5)
A solution of iron(III) acetylacetonate (0.035 g, 0.1 mmol) in DMF (2 mL) was added to a well-stirred solution of H2L (0.054 g, 0.2 mmol) in DMF (2 mL), and the resulting mixture colour changed quickly from colourless to dark brown. The mixture was further stirred at room temperature for 24 h. DMF was removed under vacuum and the obtained product of this step (typically the mononuclear species) was dissolved in THF to yield a dark solution, into which slow diffusion of methanol (during 6 to 12 h) affords well-formed orange crystals of 5·1/2CH3OH in good yield (0.045 g, 72%). IR (pure, orbit diamond, cm−1): 3513, 2814, 2921, 1619, 1588, 1520, 1487, 1364, 1252. Anal. Calc. for C46H50Fe2N8O10S8·1/2CH3OH: C, 44.37; H, 4.12; N, 8.90. Found: C, 43.98; H, 3.94; N 8.71.Synthesis of FeIII2(μ-O)(HL)4·3CH2Cl2 (6)
A mixture of iron(III) acetylacetonate (0.035 g, 0.1 mmol) and H2L (0.054 g, 0.2 mmol) in MeOH (20 mL) was stirred at room temperature in the dark and N2 for 24 h. The black precipitate observed at the end of the reaction was filtered. The obtained black powder was washed with MeOH (2 × 5 mL) and dried under vacuum. Dark green crystals suitable for X-ray diffraction were obtained by slow diffusion of diethyl ether into the CH2Cl2 solution of the black powder (yield: 0.015 g, 20%). IR (pure, orbit diamond, cm−1): 1482, 1359, 1303,1237, 979, 749. Anal. Calc. for C44H44Fe2N8O9S8·3CH2Cl2: C, 38.88; H, 3.47; N, 7.72. Found: C, 38.69; H, 3.54; N, 7.49. After the initial synthesis above, we were unable to reproduce this reaction again for further characterization.Magnetic measurements
Magnetic measurements were performed at the Institut de Physique et Chimie des matériaux de Strasbourg (UMR CNRS-ULP 7504) using a Quantum Design MPMS-XL SQUID magnetometer. The susceptibility measurement was performed in the 300–1.8 K temperature range and an applied field of 5 kOe. Isothermal field dependent magnetization measurement at room temperature confirms the absence of ferromagnetic impurities. Data were corrected for the sample holder and diamagnetism of the content estimated from Pascal constants.Crystal structure determinations
Suitable crystals for the X-ray analyses of all compounds were obtained as described above. Single crystals of 1–6 were mounted on a Nonius Kappa-CCD area detector diffractometer (Mo-Kα, λ = 0.71073 Å). The complete conditions of data collection (Denzo software40) and structure refinements are given in supporting materials. The cell parameters were determined from reflections taken from one set of 10 frames (1.0° steps in phi angle), each at 20 s exposure. The structures were solved using direct methods (SHELXS-97) and refined against F2 using the SHELXL-97 and CRYSTALBUILDER softwares.41,42 The absorption was not corrected. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were generated according to stereochemistry and refined using a riding model in SHELXL-97.Acknowledgements
We thank the CNRS, the Ministère de la Recherche (Paris) and Fundação para a Ciência e Tecnologia, (FCT), for a postdoctoral fellowship (SFRH/BPD/44262/2008) to Dr Vitor Rosa. We also thank Dr Guillaume Rogez (IPCMS – Strasbourg) for his help with magnetic measurements.References
- O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993 Search PubMed.
- A. Cornia, A. C. Fabretti, L. Zobbi, A. Caneschi, D. Gatteschi, M. Mannini and R. Sessoli, in: Single-Molecule Magnets and Related Phenomena Structure and Bonding Book Series, ed. R. Winpenny, Springer, Berlin, Heidelberg, 2006, vol. 122 pp. 133 Search PubMed.
- R. Sessoli, Mol. Cryst. Liq. Cryst., 1995, 274, 145 CrossRef.
- M. Ferbinteanu, H. Miyasaka, W. Wernsdorfer, K. Nakata, K. Sugiura, M. Yamashita, C. Coulon and R. Clérac, J. Am. Chem. Soc., 2005, 127, 3090–3099 CrossRef CAS.
- H. Hiraga, H. Miyasaka, K. Nakata, T. Kajiwara, S. Takaishi, Y. Oshima, H. Nojiri and M. Yamashita, Inorg. Chem., 2007, 46, 9661–9671 CrossRef CAS.
- C. Beghidja, G. Rogez, J. Kortus, M. Wesolek and R. Welter, J. Am. Chem. Soc., 2006, 128, 3140–3141 CrossRef CAS.
- N. Bouslimani, N. Clément, G. Rogez, P. Turek, M. Bernard, S. Dagorne, D. Martel, H. N. Cong and R. Welter, Inorg. Chem., 2008, 47, 7623–7630 CrossRef CAS.
- N. Bouslimani, N. Clément, G. Rogez, P. Turek, S. Choua, S. Dagorne and R. Welter, Inorg. Chim. Acta, 2010, 363, 213–220 CrossRef CAS.
- C. Toussaint, C. Beghidja and R. Welter, C. R. Chim., 2010, 13, 343–352 CrossRef CAS.
- N. Clément, C. Toussaint, G. Rogez, C. Loose, J. Kortus, L. Brelot, S. Choua, S. Dagorne, P. Turek and R. Welter, Dalton Trans., 2010, 39, 4579–4585 RSC.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS.
- R. Welter, WO, 2009 Search PubMed.
- C. Beghidja, M. Wesolek and R. Welter, Inorg. Chim. Acta, 2005, 358, 3881–3888 CrossRef CAS.
- M. Cheng, M. C. Chan, S. Peng, K. Cheung and C. Che, J. Chem. Soc., Dalton Trans., 1997, 3479–3482 RSC.
- E. J. Larson and V. L. Pecoraro, J. Am. Chem. Soc., 1991, 113, 3810–3818 CrossRef CAS.
- B. Li, X. Sun, G. Cheng and Z. Li, Chin. J. Chem., 2009, 27(7), 1312–1316 CrossRef CAS.
- R. Mayilmurugan, H. Stoeckli-Evans, E. Suresh and M. Palaniandavar, Dalton Trans., 2009, 5101–5114 RSC.
- G. Ilyashenko, M. Motevalli and M. Watkinson, Tetrahedron: Asymmetry, 2006, 17, 1625–1628 CrossRef CAS.
- J. Wu, S. Liu, T. D. Keene, A. Neels, V. Mereacre, A. K. Powell and S. Decurtins, Inorg. Chem., 2008, 47, 3452–3459 CrossRef CAS.
- C. J. Whiteoak, R. T. Martin de Rosales, A. J. P. White and G. J. P. Britovsek, Inorg. Chem., 2010, 49, 11106–11117 CrossRef CAS.
- K. Oyaizu, E. L. Dewi and E. Tsuchida, Inorg. Chim. Acta, 2001, 321, 205–208 CrossRef CAS.
- J. M. Becker, J. Barker, G. J. Clarkson, R. van Gorkum, G. K. Johal, R. I. Walton and P. Scott, Dalton Trans., 2010, 39, 2309–2326 RSC.
- E. Pardo, F. Lloret, R. Carrasco, M. C. Muñoz, T. Temporal-Sánchez and R. Ruiz-Garíc, Inorg. Chim. Acta, 2004, 357, 2713–2720 CrossRef CAS.
- R. Shakya, D. R. Powell and R. P. Houser, Eur. J. Inorg. Chem., 2009, 5319–5327 CrossRef CAS.
- A. K. Boudalis, N. Lalioti, G. A. Spyroulias, C. P. Raptopoulou, A. Terzis, V. Tangoulis and S. P. Perlepes, J. Chem. Soc., Dalton Trans., 2001, 955–957 RSC.
- A. K. Boudalis, C. P. Raptopoulou, A. Terzis and S. P. Perlepes, Polyhedron, 2004, 23, 1271–1277 CrossRef CAS.
- A. Bacchi, I. Ivanovic-Burmazovic, G. Pelizzi and K. Andjelkovic, Inorg. Chim. Acta, 2001, 313, 109–119 CrossRef CAS.
- H. Chun, C. N. Verani, P. Chaudhuri, E. Bothe, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 2001, 40, 4157–4166 CrossRef CAS.
- S. Mukherjee, T. Weyhermüller, E. Bill, K. Wieghardt and P. Chaudhuri, Inorg. Chem., 2005, 44, 7099–7108 CrossRef CAS.
- B. Li, J. Zhang, X. Zhang, J. Tian and G. Huang, Inorg. Chim. Acta, 2011, 366, 241–246 CrossRef CAS.
- N. Bouslimani, N. Clément, C. Toussaint, S. Hameury, P. Turek, S. Choua, S. Dagorne, D. Martel and R. Welter, Eur. J. Inorg. Chem., 2009, 3734–3741 CrossRef CAS.
- G. Ilyashenko, M. Motevalli and M. Watkinson, Tetrahedron: Asymmetry, 2006, 17, 1625–1628 CrossRef CAS.
- D. M. Kurtz, Jr., Chem. Rev., 1990, 90, 585–606 CrossRef.
- Q. Meng, L. Wang, Y. Liu and Y. Pang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m63 Search PubMed.
- A. Ozarowski, B. R. McGarvey and J. E. Drake, Inorg. Chem., 1995, 34, 5558–5566 CrossRef CAS.
- G. L. Parrilha, C. Fernandes, A. J. Bortoluzzi, B. Szpoganicz, M. de S. Silva, C. T. Pich, H. Terenzi and A. Horn, Jr., Inorg. Chem. Commun., 2008, 11, 643–647 CrossRef CAS.
- T. Kojima, R. A. Leising, S. Yan and L. Que, Jr., J. Am. Chem. Soc., 1993, 115, 11328–11335 CrossRef CAS.
- R. M. Buchanan, S. Chen, J. F. Richardson, M. Bressan, L. Forti, A. Morvillo and R. H. Fish, Inorg. Chem., 1994, 33, 3208–3209 CrossRef CAS.
- J. Xu, J. Astner, O. Walter, F. W. Heinemann, S. Schindler, M. Merkel and B. Krebs, Eur. J. Inorg. Chem., 2006, 1601–1610 CrossRef CAS.
- B. V. Nonius, Kappa CCD Operation Manual, Delft, The Netherlands, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- R. Welter, Acta Crystallogr., Sect. A: Found. Crystallogr., 2006, 62, s252 Search PubMed.
Footnote |
| † CCDC reference numbers 844919–844924. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra01316a |
| This journal is © The Royal Society of Chemistry 2012 |