Liesegang rings of dendritic silver crystals emerging from galvanic displacement reaction in a liquid-phase solution†
Shunping
Xie
ab,
Xicui
Zhang
a,
Suolong
Yang
c,
Man Chin
Paau
b,
Dan
Xiao
*a and
Martin M. F.
Choi
*b
aCollege of Chemistry and College of Chemical Engineering, Sichuan University, Chengdu 610064, People's Republic of China. E-mail: xiaodan@scu.edu.cn; Fax: +86-28-85416029; Tel: +86-28-85415029
bDepartment of Chemistry, Hong Kong Baptist University, 224 Waterloo Road, Kowloon Tong, Hong Kong SAR, People's Republic of China. E-mail: mfchoi@hkbu.edu.hk; Fax: +852 34117348; Tel: +852-34117839
cScience and Technology on Surface Physics and Chemistry Laboratory, P.O. Box 718-35, Mianyang 621907, People's Republic of China
First published on 23rd March 2012
Abstract
Liesegang rings (LR) of various types of Ag dendrite crystals are observed from an aqueous solution based on a simple galvanic displacement reaction between a Zn plate and Ag2SO4. The growth process and formation mechanism of LR of Ag dendrites are investigated in detail. The synthesised LR of Ag dendrites have been successfully employed as a Raman probe for SERS analysis of melamine.
Liesegang rings (LR) with their own unique scenery of periodic precipitation bands/rings have intrigued scientists for over a century and stimulated the launch of extensive experiments and theoretical studies.1 In a typical experiment of a developing LR pattern, an electrolyte (inner electrolyte) is uniformly loaded in the reaction medium (usually a gel) while another electrolyte (outer electrolyte) diffuses from outside into the reaction medium. A sparingly soluble or insoluble immobile product with a rhythmic pattern of precipitates parallel to the front of the diffusing electrolyte is thus formed as a result of diffusion and precipitation reaction. Actually, the nature of precipitation does not play an important role in the onset mechanism. The gel may influence the structural details but is not essential for pattern formation. It also prevents convection and sedimentation of the solid phase. A number of models explaining the formation of LR patterns have been developed and they can be summarised as two main theories: prenucleation2 and postnucleation.3
LR phenomena are also found in a wide variety of natural biological and geological systems such as bacteria colonies, gallstones, plants, tissues,4 agate rocks, concretions, geodes, and orbicules.5 These findings are conducive to our better understanding of the various physiochemical processes involved in the formation mechanism of specific patterns. In addition, LR can be applied to determine sulfate concentration in individual rain droplets6 and to identify atmospheric aerosol particles.7
The formation of LR patterns is mainly described as the interaction and diffusion of the ionic species in a gel medium.1d,8 So far the generation of LR patterns from nanoparticles (NPs) have not received much attention. Lagzi et al.9 first reported that functionalised, oppositely charged gold nanoparticles (AuNPs) or silver nanoparticles (AgNPs) could give rise to self-organisation of LR. The formation of the LR of NPs aggregates on a 2 mm thick sheet of 0.5 wt% agarose gel system is attributed to the electrostatic attraction and stabilisation of the positively and negatively charged AuNPs. Traditionally the ionic species act as building blocks for LR patterns via their self-organisation and co-precipitation. To date nanoscopic components can also be engineered for the formation of LR in a gel medium. To our knowledge, almost all formations of LR are dependent on coupling precipitation with diffusion in a gel medium. There is still a need to explore other simpler types of reaction systems such as aqueous solution so that a wider range of nanomaterials can be employed to create LR patterns. Moreover, it will be relatively simple and easy to control and manipulate the formation of LR in aqueous solution.
To date, many nanostructures exhibiting unique and tuneable properties have been synthesised by galvanic exchange reaction. Herein, we report for the first time the LR patterns of various types of Ag dendrite crystals prepared from aqueous solution systems based on a galvanic displacement reaction. The formation of LR of Ag dendrite crystals is attributed to the concentration gradient and diffusion of electrolytes in aqueous solution. We anticipate that our work can attract considerable interests in designing more LR patterns in aqueous solutions. In addition, melamine has been chosen as the model compound to demonstrate that the Ag dendrites in LR is a very useful solid substrate for surface enhanced Raman spectroscopy (SERS). This communication illustrates a very simple and fast route to synthesise LR of Ag dendrites which is potentially useful in SERS. Finally, the growth of the LR of Ag dendrites can be seen visually and has been real-time recorded by video.
The whole LR experiment involves readily available materials: a zinc (Zn) plate (> 99%, Tianjin No. 3 Chemical Reagent Factory, China) and a silver sulfate (Ag2SO4, Tianjin Kermel Chemical Reagents, China) aqueous solution. In a typical experiment, the Zn plate (diameter 6 mm) was firstly treated with dilute sulfuric acid, washed with doubly deionised distilled (DDI) water, and finally sonicated in DDI water for 2 min to remove the surface debris and impurities. The pre-treated Zn plate was immersed into a 6.0 cm diameter glass Petri dish containing 14 mL of 5.0 mM Ag2SO4 aqueous solution.
Fig. 1 displays the photographic images of the LR of dendritic Ag crystals formed at various reaction times. At the beginning, the first ring does not grow evenly around the Zn plate but transforms into a more ring-like structure at the reaction time of 10–30 min. As the reaction time proceeds, white–grey, grey, and dark grey LR are alternatively grown outward. The longer the reaction time, the more LR are formed. In general, the rings changes from the initial dark grey to pale grey and finally to whitish grey with prolonged standing. Some of the rings even turn into a Ag mirror which will be discussed later.
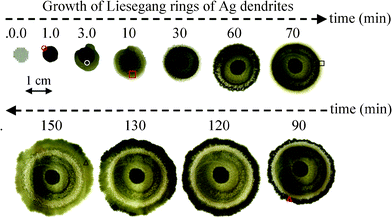 | ||
| Fig. 1 Photographic images of Liesegang rings of dendritic Ag crystals taken at various reaction times. The Zn plate is in the centre of the rings and LR of dendritic Ag crystals grows around the Zn plate. | ||
In a galvanic exchange, a metal in the reduced form is replaced by another metal in the oxidised form by a simple redox reaction. The appearance of our LR can be explained by the very rapid galvanic displacement reaction between Zn atoms of the plate and Ag+ ions in the solution: Zn(s) + 2Ag+(aq) → Zn2+(aq) + 2Ag(s). Since the standard electrode potential of the Ag+/Ag pair (EoAg+/Ag = +0.799 V) is larger than that of Zn2+/Zn pair (EoZn2+/Zn = −0.762 V), the driving force for the galvanic replacement reaction is the electrical potential difference between the Ag+/Ag and Zn2+/Zn pairs. For instance, metal A of lower reduction potential can displace a metal ion B+ from its salt solution (BX) when metal B has a higher reduction potential (A + BX → AX + B, where X is an anion). Galvanic displacement reaction does not require an external reducing agent or electric current. The electrons from the valence band or bonding electrons of the solid material can spontaneously reduce metal ions in solution to metallic particles. Once the Zn plate is in contact with the Ag2SO4 aqueous solution, the spontaneous redox reaction between Zn metal and Ag2SO4 aqueous solution will occur on the surface and/or edge of the Zn plate.
The Zn plate tends to lose its electrons and dissolves in the aqueous solution as Zn2+ ions and simultaneously the Ag+ ions in the solution accept the electrons from the Zn plate and then deposit as AgNPs on the surface and/or edge of the Zn plate. When the Zn plate is positioned into a 5.0 mM Ag2SO4 aqueous solution for 1 min, the whole surface or edge of the Zn plate is covered by black aggregated AgNPs. The SEM image clearly indicates that the Zn plate is densely occupied by aggregated AgNPs in the first 1 min (Fig. S1 of ESI†). As time passes by, more AgNPs are deposited to build up the nano-sized primary electrolytic cells. That is to say, these electrolytic cells comprising a Zn anode and Ag cathode are set up in close proximity to the Zn plate. The redox reaction between Zn atoms and Ag+ ions generate electric currents that support the continuous fast growth of cathodic AgNPs into Ag dendrites. The reaction mechanisms are illustrated in Fig. 2. These AgNPs spontaneously evolve to dense Ag dendrites after about 1 min, revealed by the SEM image (Fig. S2†).
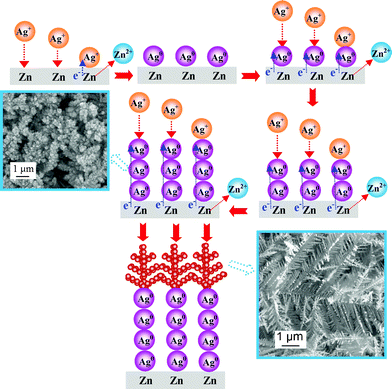 | ||
| Fig. 2 Reaction mechanisms illustrate the formation of Liesegang rings of Ag dendrites (not drawn to scale). | ||
The subsequent growth of Ag dendrites would preferentially take place on the newly formed AgNPs through a primary cell reaction rather than on the bare Zn surfaces or edges via surface reaction. Ag+ ions in the bulk solution move through the diffusion layer under the effect of a concentration gradient to reach the AgNP reactive sites of the dendrites, where reduction of Ag+ to Ag0 could take place spontaneously. As time passes by, more Ag+ ions are rapidly deposited on the existing AgNPs which will evolve into the final fractal Ag dendrites under the non-equilibrium or kinetically controlled condition.10 Subsequently the Ag dendrites coalesce with neighbouring dendrites to form a Ag mirror structure to lower its surface energies under thermodynamically controlled conditions. The whole growth process of the LR of Ag dendrites and mirrors can be observed in real-time and recorded by a LCD video zoom microscope (Video 1 in ESI†).
At the bottom of the Petri dish, the Ag mirror stops growing after 10 min. The Ag mirror is derived from a lot of dendritic Ag crystals which are originally the aggregated AgNPs (Fig. S3†). Various dendritic Ag structures are also formed on the surface of this Ag mirror as depicted in Fig. 3.
 | ||
| Fig. 3 SEM images of different types (A)–(F) of dendritic Ag structure deposited on the surface of Ag mirror emerging from Zn plate obtained in 5.0 mM Ag2SO4 aqueous solution at a reaction time of 10 min. SEM images were captured in the areas indicated by the red rectangle in Fig. 1. | ||
These Ag dendrites differ mainly by the number of branching generations and the angles between stems and branches. Fig. 3A–C displays the Ag branches which are flanked by stems symmetrically at an angle of ∼60°. The two-generation dendritic structure is very sparse in Fig. 3A and C as compared to Fig. 3B. The angles between the stems and branches in Fig. 3D and E are all 90°. In addition, some aggregated AgNPs are grown on the stems or side branches of the dendritic Ag structure (Fig. 3A and F). Large Ag slices are even attached to some Ag dendrites as shown in Fig. S3.†
In the first 10 min, the growth of Ag dendrites is very fast due to the initial high concentration of Ag2SO4 (5.0 mM), observed under a real-time microscope video recorder (Video 2 in ESI†). As the reaction proceeds from 10 to 30 min, the Ag+ ion gradually exhausts, resulting in the continuous slow growth of Ag dendrites. In the diffusion layer, the concentration of Ag+ ion drops sharply to a level that the galvanic displacement reaction becomes slower. The Ag dendrites in this ring will subsequently rearrange and transform to larger shiny Ag dendrites, i.e., from the non-equilibrium to the quasi-equilibrium and then to equilibrium conditions. In other words, the Ag dendrites may have enough time to relax and transfer to the minimum energy position, leading to a thermodynamically stable shape10 (Fig. S4†). In essence, the growth of black Ag dendrites is a little earlier than the shiny Ag dendrites. A kinetic factor driven by the concentration gradient dominates the growth processes of AgNPs and Ag dendrites.11
When the reaction continues from 30 to 60 min, there are sufficient Ag+ ions from the bulk solution to diffuse to the previous LR and deposit as black AgNPs, generating the next LR. Again, the galvanic displacement reaction gradually drops as the Ag+ ions in the diffusion layer close to this LR diminish. At this moment, these small black Ag dendrites rearrange to be shiny Ag dendrites. As the reaction time increases to 70 min, all the black Ag dendrites evolve into larger shiny Ag dendrites with thermodynamically stable shape which is similar to that depicted in Fig. S4.† More LR are generated via the same cycle. In summary, the formation of LR is governed by the Ag+ ions in the diffusion layer between each ring. A schematic diagram to illustrate the reaction–diffusion of Ag+ and formation of LR is displayed in Fig. S5.†
When the reaction time reaches 90 min, black Ag dendrites with both perfect and imperfect dendritic structures are obtained (Fig. S6†). After 150 min, the Ag dendrites changes from black to grey attributing to the attachment of some Ag slices on the black Ag dendrites (Fig. S7†). All the SEM images in the ESI† are the newly formed rings at various reaction times.
Finally, the whole LR pattern is formed, of which one ring (ring 1) is a Ag mirror covered by Ag dendrites, two rings (rings 2 and 4) are shiny stable structures of larger Ag dendrites attached with some smaller Ag dendrites, and two rings (rings 3 and 5) with different morphologies of small Ag dendrites as shown in Fig. S8(A).† The LR (except ring 1) obey the spacing law of successive ring locations:9,12xn+1/xn = p, where xn+1 and xn are the distance of n+1 and n rings from the zinc plate respectively, and p denotes a constant spacing coefficient as depicted in Table 1. The spacing coefficient is found to be p = 1.3.
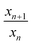
Fig. S8(A)† displays the LR formed at the top layer of the Petri dish, which is more greyish than that of the bottom layer. In fact, the Ag dendrites at the top layer can continuously encounter fresh Ag+ ions whereas the bottom layer cannot. As such, once the Ag dendrites are formed at the bottom layer and no more fresh Ag+ ions are available, these Ag dendrites will have more time to rearrange to large shiny Ag mirror plates to minimise their surface energies (Fig. S4†).
Fig. S8(B) and (C)† display the XRD and XPS spectra of the Ag dendrites at different rings and all are confirmed to be elemental Ag crystals. It should be noted that LR cannot be formed if the initial concentration of the Ag2SO4 solution is either too low (<1 mM) or too high (>10 mM). At low concentrations, only a Ag mirror with a few AgNPs is obtained. By contrast, when the Ag2SO4 concentration is too high, large amounts of grey Ag dendrites on shiny Ag dendrites are obtained. As such, 5.0 mM Ag2SO4 is chosen as the optimal initial concentration since it can produce LR as depicted in Fig. 1.
Raman spectroscopy has emerged as a fast, non-invasive analytical method for the detection and quantification of adulterants in many fields. In general, conventional Raman spectroscopy produces weak signals but this can be greatly improved by SERS. Many SERS-active substrates such as NPs,13 nanoshells,14 nanorods,15 nanotubes,16, nanoplates,17 nanoprisms,18 nanowires19 and dendrites20 have been developed. In recent years, Ag dendritic nanostructures have attracted considerable interests due to their large surface area and intriguing hierarchical structure sprawling to several generations. To explore the potential application of our Ag dendrites, SERS was applied to study melamine. An aliquot of 10 mL of 0.10 mM melamine aqueous solution was added to a Petri dish with LR of Ag dendrites and left for 24 h. Then, the solution was discarded and the LR of Ag dendrites in the Petri dish were air-dried at room temperature. Afterwards, the melamine-adsorbed Ag dentrites were analysed by Raman spectroscopy. A blank Petri dish without LR of Ag dendrites is used for comparison. An excitation light beam of 532 nm from an air-cooled argon ion laser source operating at 25 mW was focused onto various spots of the sample on a microscope stage through a 10× objective. The Raman spectra were obtained from averaging four successive scans with 4 s integration time in the 500–1700 cm−1 region. Fig. 4 displays the Raman spectra obtained from (a) solid melamine, (b) melamine on a blank Petri dish, (c) melamine adsorbed on larger shiny Ag dendrites in the Petri dish (marked with red at the bottom view of LR in Fig. S8(A)†), and (d) melamine adsorbed on smaller Ag dendrites (marked with blue at the top view of LR in Fig. S8(A)†).
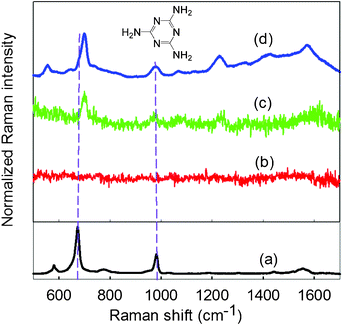 | ||
| Fig. 4 Raman spectra of (a) solid melamine, (b) melamine on a blank Petri dish, (c) melamine adsorbed on larger shiny Ag dendrites, and (d) melamine adsorbed on smaller Ag dendrites. Measurement was conducted from 500 to 1700 cm−1 with 4 s exposure time and 25 mW laser power. The chemical structure of melamine is displayed at the top. | ||
Typical Raman peaks of solid melamine at 676 and 984 cm−1 are identified from Fig. 4a. The most intense peak at 676 cm−1 is assigned to the ring-breathing II mode, involving the in-plane deformation of the triazine ring. Another strong peak at 984 cm−1 arises from the ring-breathing mode I of the triazine ring.21 No peaks are observed in the Raman spectrum of the blank Petri dish (Fig. 4b). By contrast, stronger Raman signals can be obtained using Ag dendrites as the SERS substrate. It was also found that the Raman signals obtained from the smaller Ag dendrites (Fig. 4d) are much stronger than that of the larger shiny Ag dendrites (Fig. 4c). The melamine peak at 699 is shifted by 23 cm−1, which is a large shift compared to that of the solid melamine, attributing to the electromagnetic field and chemical or electronic enhancement effect of Ag dendrites on the triazine ring of melamine.22 Clearly, the relative intensities of the Raman peaks at 699 and 984 cm−1 are much enhanced for smaller Ag dendrites. Here, the as-synthesised Ag dendrites indeed enhance the Raman signal of melamine, providing a useful approach to the qualitative determination of melamine.
Conclusion
In summary, LR of dendritic Ag emerging from galvanic displacement reaction in liquid-phase solution are first observed. The formation of LR of Ag dendrite crystals is attributed to the concentration gradient and diffusion of Ag+ ions in aqueous solution. We anticipate that our results will provoke more discoveries of other LR patterns in aqueous solution systems. Furthermore, the as-synthesised LR of Ag dendrites have been successfully employed as a Raman probe for SERS analysis of melamine. Our work provides a very simple, convenient, cost-effective, and fast route to synthesise LR of Ag dendrites which is potentially useful in SERS and other applications.Financial support from the HKBU Faculty Research Grant (FRG2/08–09/077) is gratefully acknowledged. M. C. Paau acknowledges the receipt of a postgraduate studentship from the Additional UGC-funded Research Postgraduate Places in the area of Environmental and Human Health Risk Assessment of Persistent Toxic Substances.
References
- (a) T. Karam, H. El-Rassy and R. J. Sultan, J. Phys. Chem. A, 2011, 15, 2994 CrossRef; (b) L. Badr, Z. Moussa, A. Hariri and R. Sultan, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2011, 83, 016109(1) CrossRef; (c) I. Lagzi and D. Ueyama, Chem. Phys. Lett., 2009, 468, 188 CrossRef CAS; (d) M. Bhattacharya, D. G. Vlachos and M. Tsapatsis, Appl. Phys. Lett., 2003, 82, 3357 CrossRef CAS; (e) S. C. Müller and J. Ross, J. Phys. Chem. A, 2003, 107, 7997 CrossRef; (f) I. T. Bensemann, M. Fialkowski and B. A. Grzybowski, J. Phys. Chem. B, 2005, 109, 2774 CrossRef CAS; (g) B. A. Grzybowski, K. J. M. Bishop, C. J. Campbell, M. Fialkowski and S. K. Smoukov, Soft Matter, 2005, 1, 114 RSC.
- (a) S. Prager, J. Chem. Phys., 1956, 25, 279 CrossRef CAS; (b) D. A. Smith, J. Chem. Phys., 1984, 81, 3102 CrossRef CAS.
- (a) R. Feeney, L. S. Schmidt, P. Strickholm, J. Chadam and P. Ortoleva, J. Chem. Phys., 1983, 78, 1293 CrossRef CAS; (b) G. Venzl, J. Chem. Phys., 1986, 85, 1996 CrossRef CAS.
- D. Xie, J. Wu, G. Xu, Q. Ouyang, R. D. Soloway and T. Hu, J. Phys. Chem. B, 1999, 103, 8602 CrossRef CAS.
- (a) F. Marko, D. Pivko and V. Hurai, Geol. Q., 2003, 47, 241 Search PubMed; (b) P. J. Heaney and A. M. Davis, Science, 1995, 269, 1562 CAS.
- K. Narita, K. Matsumoto and M. Igawa, Anal. Sci., 2006, 22, 1559 CrossRef CAS.
- K. R. Spurny, Analytical chemistry of aerosols, Lewis Publishers, Boca Raton, 1999, pp. 231–242 Search PubMed.
- (a) T. Narita and M. Tokita, Langmuir, 2006, 22, 349 CrossRef CAS; (b) L. Mandalian, M. Fahs, M. Al-Ghoul and R. Sultan, J. Phys. Chem. B, 2004, 108, 1507 CrossRef CAS; (c) M. I. Lebedeva, D. G. Vlachos and M. Tsapatsis, Phys. Rev. Lett., 2004, 92, 088301(1) CrossRef.
- I. Lagzi, B. Kowalczyk and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 58 CrossRef CAS.
- (a) J. Fang, H. You, P. Kong, Y. Yi, X. Song and B. Ding, Cryst. Growth Des., 2007, 7, 864 CrossRef CAS; (b) R. Liu and A. Sen, Chem. Mater., 2012, 24, 48 CrossRef CAS.
- J. Fang, H. You, C. Zhu, P. Kong, M. Shi, X. Song and B. Ding, Chem. Phys. Lett., 2007, 439, 204 CrossRef CAS.
- K. Jablczynski, Bull. Soc. Chim. Fr., 1923, 33, 1592 Search PubMed.
- (a) S. Preciado-Flores, D. A. Wheeler, T. M. Tran, Z. Tanaka, C. Jiang, M. Barboza-Flores, F. Qian, Y. Li, B. Chen and J. Z. Zhang, Chem. Commun., 2011, 47, 4129 RSC; (b) R. G. Freeman, K. C. Grabar, K. J. Allison, R. M. Bright, J. A. Davis, A. P. Guthrie, M. B. Hommer, M. A. Jackson, P. C. Smith, D. G. Walter and M. J. Natan, Science, 1995, 267, 1629 CAS; (c) L. Rivas, S. Sanchez-Cortes, J. V. García-Ramos and G. Morcillo, Langmuir, 2000, 16, 9722 CrossRef CAS; (d) M. M. Maitani, D. A. A. Ohlberg, Z. Li, D. L. Allara, D. R. Stewart and R. S. Williams, J. Am. Chem. Soc., 2009, 131, 6310 CrossRef CAS; (e) J. Vongsvivut, E. G. Robertson and D. McNaughton, J. Raman Spectrosc., 2010, 41, 1137 CrossRef CAS.
- (a) L. Lu, H. Zhang, G. Sun, S. Xi, H. Wang, X. Li, X. Wang and B. Zhao, Langmuir, 2003, 19, 9490 CrossRef CAS; (b) K. N. Heck, B. G. Janesko, G. E. Scuseria, N. J. Halas and M. S. Wong, J. Am. Chem. Soc., 2008, 130, 16592 CrossRef CAS; (c) A. Barhoumi, D. Zhang and J. Halas, J. Am. Chem. Soc., 2008, 130, 14040 CrossRef CAS.
- (a) B. Nikoobakht and M. A. El-Sayed, J. Phys. Chem. A, 2003, 107, 3372 CrossRef CAS; (b) Y.-J. Liu, Z.-Y. Zhang, R. A. Dluhy and Y.-P. Zhao, J. Raman Spectrosc., 2010, 41, 1112 CrossRef CAS; (c) S. Mohapatra, S. Siddhanta, D. R. Kumar, C. Narayana and T. K. Maji, Eur. J. Inorg. Chem., 2010, 4969 CrossRef CAS.
- A. Roguska, A. Kudelski, M. Pisarek, M. Opara and M. Janik-Czachor, Appl. Surf. Sci., 2011, 257, 8182 CrossRef CAS.
- (a) X. Huang, S. Tang, B. Liu, B. Ren and N. Zheng, Adv. Mater., 2011, 23, 3420 CrossRef CAS; (b) Y. Sun and G. P. Wiederrecht, Small, 2007, 3, 1964 CrossRef CAS.
- J. Y. Xu, J. Wang, L. T. Kong, G. C. Zheng, Z. Guo and J. H. Liu, J. Raman Spectrosc., 2011, 42, 1728 CrossRef CAS.
- (a) P. Mohanty, I. Yoon, T. Kang, K. Seo, K. S. K. Varadwaj, W. Choi, Q.-H. Park, J. P. Ahn, Y. D. Suh, H. Ihee and B. Kim, J. Am. Chem. Soc., 2007, 129, 9576 CrossRef CAS; (b) N. L. Netzer, C. Qiu, Y. Zhang, C. Lin, L. Zhang, H. Fong and C. Jiang, Chem. Commun., 2011, 47, 9606 RSC; (c) S. Preciado-Flores, D. A. Wheeler, T. M. Tran, Z. Tanaka, C. Jiang, M. Barboza-Flores, F. Qian, Y. Li, B. Chen and J. Z. Zhang, Chem. Commun., 2011, 47, 4129 RSC.
- (a) S. Xie, X. Zhang, D. Xiao, M. C. Paau, J. Huang and M. M. F. Choi, J. Phys. Chem. C, 2011, 115, 9943 CrossRef CAS; (b) A. Gutés, C. Carraro and R. Maboudian, J. Am. Chem. Soc., 2010, 132, 1476 CrossRef; (c) X. Chen, C.-H. Cui, Z. Guo, J.-H. Liu, X.-J. Huang and S.-H. Yu, Small, 2011, 7, 858 CrossRef CAS; (d) L. He, T. Rodda, C. L. Haynes, T. Deschaines, T. Strother, F. Diez-Gonzalez and T. P. Labuza, Anal. Chem., 2011, 83, 1510 CrossRef CAS; (e) J. Huang, S. Vongehr, S. Tang, H. Lu, J. Shen and X. Meng, Langmuir, 2009, 25, 11890 CrossRef CAS; (f) G. Lu, C. Li and G. Shi, Chem. Mater., 2007, 19, 3433 CrossRef CAS; (g) D. K. Sharma, A. Ott, A. P. O'Mullane and S. K. Bhargava, Colloids Surf., A, 2011, 386, 98 CrossRef CAS; (h) L. He, M. Lin, H. Li and N.-J. Kim, J. Raman Spectrosc., 2010, 41, 739 CAS.
- E. Koglin, B. J. Kip and R. J. Meier, J. Phys. Chem., 1996, 100, 5078 CrossRef CAS.
- X.-F. Zhang, M.-Q. Zou, X.-H. Qi, F. Liu, X.-H. Zhu and B.-H. Zhao, J. Raman Spectrosc., 2010, 41, 1655 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: details of characterization methods for Ag dendrites, supplementary figures of SEM, XRD and XPS of various nanostructures of dendritic Ag crystals, schematic diagram illustrating the reaction–diffusion and formation process of LR, and growth videos of LR of Ag dendrites with Video 1 as the normal play time and Video 2 as 10 times faster than the normal play time. See DOI: 10.1039/c2ra20055d/ |
| This journal is © The Royal Society of Chemistry 2012 |