Direct growth of FeVO4 nanosheet arrays on stainless steel foil as high-performance binder-free Li ion battery anode†
Dao
Hao Sim‡
a,
Xianhong
Rui‡
ab,
Jing
Chen
a,
Huiteng
Tan
a,
Tuti Mariana
Lim
bc,
Rachid
Yazami
a,
Huey Hoon
Hng
*a and
Qingyu
Yan
*ade
aSchool of Materials Science and Engineering, Nanyang Technological University, 639798, Singapore. E-mail: ashhng@ntu.edu.sg; alexyan@ntu.edu.sg
bSchool of Civil and Environmental Engineering, Nanyang Technological University, 639798, Singapore
cSchool of Life Sciences and Chemical Technology, Ngee Ann Polytechnic, 599489, Singapore
dEnergy Research Institute, Nanyang Technological University, 637459, Singapore
eTUM CREATE Centre for Electromobility, Nanyang Technological University, 637459, Singapore
First published on 21st February 2012
Abstract
Amorphous FeVO4 nanosheet arrays have been grown directly from a flexible stainless steel (SS) substrate by a facile template-free and catalyst-free chemical vapour deposition (CVD) method. These FeVO4 nanosheets showed superior Li storage properties, especially at high current densities.
Transition metal oxides have been studied extensively as Li ion battery (LIB) anodes due to their chemical stability, low cost and being environmental friendly. Among them, metal vanadates are a very interesting group of materials. Although the theoretical capacities are not clearly reported, several reports have demonstrated that very high specific capacities can be achieved in metal vanadates,1–5e.g., ∼1300 mAh g−1 for FeVO4 and >1000 mAh g−1 for InVO4. These values are considerably higher than those achieved in other oxides that have attracted great attention so far (e.g. ∼960 mAh g−1 for Fe2O3 and 780 mAh g−1 for SnO2). More interestingly, it has been reported that amorphous metal vanadates deliver higher specific capacities as compared to crystallized ones.1 However, the reports on metal vanadates for LIBs anodes are relatively limited, which is mainly due to the fast capacity fading upon charge–discharge cycling. For example, it has been shown that amorphous FeVO4 depicted an initial specific capacity of ∼1300 mAh g−1, which decreased to ∼800 mAh g−1 after 10 cycles.1 Downsizing materials to the nanoscale level could be an effective approach to develop high-performance LIBs due to increased electrode–electrolyte contact area and shortened Li+ ion diffusion paths.6–9 It is thus promising to prepare nanostructured metal vanadates (e.g. FeVO4) and also maintain them as amorphous structures for LIB anode applications.
Recently, significant improvements in battery performance have been achieved by directly growing self-supported one-dimensional (1D) or two-dimensional (2D) nanostructure arrays on current-collecting substrates as anodes,10–16 which allows efficient charge transport and fast Li+ diffusion (Fig. S1, ESI†). One advantage is that the need for polymer binders and conductive carbon, which add extra weight, is eliminated. Herein, we report a facile template-free and catalyst-free chemical vapour deposition (CVD) method for the growth of amorphous FeVO4 nanosheet arrays on stainless steel (SS) foils. Here, the SS foil served as both the Fe source and the substrate/current collector for the deposition of vanadium and oxygen vapour. As an anode material for LIBs, the as-grown FeVO4 nanosheet arrays can deliver high specific capacities with a stable cycling performance, especially at high C rates, e.g., delivering reversible capacities of 601 and 453 mAh g−1 at high current densities of 10 (8 C) and 20 A g−1 (15 C), respectively.
The inset of Fig. 1a shows a typical optical image of the FeVO4 film (yellowish brown color) uniformly grown on a SS substrate collected at a substrate distance (dsub) of 10 cm away from the center of the tube furnace with a substrate temperature of TS = 673 K, which is judged from the temperature profile of the furnace at 723 K (Fig. S2, ESI†). The sample of FeVO4 on the SS foil can be rolled up without any visible signs of degradation, implying a possible application of the product in flexible electronics. Top-view FESEM images (Fig. 1a–b) demonstrate that uniform networks of interconnected nanosheets are vertically aligned on the SS substrate. The width and thickness of the nanosheets are in the ranges of 100–500 and 10–40 nm, respectively. The open space between the neighboring nanosheets is up to several hundred nm. From the side-view of the nanosheet arrays (Fig. 1c), the thickness of the as-grown FeVO4 film is ∼4 μm.
 | ||
| Fig. 1 (a) and (b) Top-view FESEM images of the as-grown film on a SS substrate collected at dsub = 10 cm. Inset in (a): corresponding optical image of the as-prepared sample on a SS foil. (c) Side-view FESEM image of the film, showing a thickness of ∼4 μm. (d) TEM image of a single nanosheet and corresponding SAED pattern (inset). | ||
The crystal structure of the as-grown film is examined by X-ray diffraction (XRD). It is shown that (Fig. S3, ESI†) there are no obvious peaks corresponding to iron oxides (e.g., FeO, Fe3O4 and Fe2O3) and iron vanadates (e.g., FeVO4 and Fe2V4O13), indicating the product is not well crystallized. This amorphous behaviour is confirmed by the selected area electron diffraction (SAED) pattern of a single nanosheet (Fig. 1d). The EDX measurement in the TEM reveals the presence of Fe, V and O, and these elements are homogeneously dispersed in the nanosheet (Fig. S4a–d, ESI†). The Cu and C signals in Fig. S4a (ESI†) originate from the TEM grids. Quantitative analysis using inductively coupled plasma (ICP) spectroscopy shows the atomic ratio of Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :V = 1:1, which matches the ratio of Fe to V in FeVO4. The oxidation states of Fe and V were determined by X-ray photoelectron spectroscopy (XPS) analysis. Since the analysis depth of the XPS technique is about a few nanometres, our film (∼4 μm thickness) is thick enough to ensure that all detected signals are from the as-prepared sample. The Fe 2p spectrum (Fig. S4e, ESI†) shows that the binding energies of Fe 2p3/2 and Fe 2p1/2 are located at 711.0 and 724.8 eV, respectively, which agree well with that reported for Fe3+.17 Fig. S4f (ESI†) shows the XPS V 2p spectrum containing two peaks of V 2p3/2 (516.9 eV) and V 2p1/2 (524.5 eV), which identifies the typical characteristics of V5+.18 On the basis of the above results, the final product is proposed to be amorphous FeVO4.
:V = 1:1, which matches the ratio of Fe to V in FeVO4. The oxidation states of Fe and V were determined by X-ray photoelectron spectroscopy (XPS) analysis. Since the analysis depth of the XPS technique is about a few nanometres, our film (∼4 μm thickness) is thick enough to ensure that all detected signals are from the as-prepared sample. The Fe 2p spectrum (Fig. S4e, ESI†) shows that the binding energies of Fe 2p3/2 and Fe 2p1/2 are located at 711.0 and 724.8 eV, respectively, which agree well with that reported for Fe3+.17 Fig. S4f (ESI†) shows the XPS V 2p spectrum containing two peaks of V 2p3/2 (516.9 eV) and V 2p1/2 (524.5 eV), which identifies the typical characteristics of V5+.18 On the basis of the above results, the final product is proposed to be amorphous FeVO4.
In order to understand the growth process of FeVO4 nanosheet arrays, a series of experiments were carried out. It is found that the bare SS foil is relatively smooth (Fig. S5a–b, ESI†). After annealing the SS foil at 673 K in air for 30 min, it becomes rather rough and some nanosheet arrays emerge on the SS foil (Fig. S5c, ESI†), which were confirmed to be α-Fe2O3 (JCPDS no. 33-0664) by SAED pattern analysis (Fig. S5d, ESI†). It is thus expected that these α-Fe2O3 can act as seeds to initiate the growth of the FeVO4 nanosheets. In addition, the substrate temperature (i.e., substrate distance) in our CVD system was noted to affect the sample morphology. Increasing the substrate temperature to ∼713 K (dsub = 4 cm) led to the formation of 100–400 nm irregular-shaped particles (Fig. S6a, ESI†). This is mainly attributed to the fast surface diffusion of the as-deposited atoms along the substrate surface. At dsub = 7 cm (TS = ∼696 K), mixed nanostructures of the particles and nanosheets were observed (Fig. S6b–d, ESI†). Uniform nanosheet arrays on SS foils were achieved at dsub = 10 cm (Fig. 1a–b). Further decreasing TS (e.g., at dsub = 15 cm) results in the reduced density of nanosheets (Fig. S6e, ESI†). At dsub > 15 cm, there was no observable growth of sheets on the SS foil.
To study the Li-ion storage properties of the amorphous FeVO4 nanosheet arrays, a series of electrochemical measurements were carried out based on the half cell configuration.7 The FeVO4 nanosheet arrays on the SS substrate were directly applied as the working electrode without addition of any other ancillary materials such as carbon black or polymer binder. Galvanostatic charge–discharge cycling was carried out in the voltage window of 0.005–3.5 V (vs. Li/Li+) at a current density of 200 mA g−1 (0.15 C, here 1 C = 1300 mA g−1) for up to 100 cycles at ambient temperature (296 K), and the voltage versus capacity/lithium number (x) profiles for the first two cycles are shown in Fig. 2a. During the first discharge (Li+ intercalation), the voltage quickly decreases to ∼0.85 V, and a capacity of about 465 mAh g−1 is delivered. This corresponds to an intercalation of 3.0 Li per unit formula of FeVO4. Following this, a gradual drop in voltage takes place until the end of discharge with a specific capacity up to 1228 mAh g−1 (consumption of 10.8 Li per unit formula of FeVO4). This large capacity obtained at the low voltage region includes the contribution from the formation of a solid electrolyte interface (SEI) and the polymeric gel-type layer on the surface of the active materials.19 The above electrochemical behaviour differs from the reported discharge curves of Fe(III) and V(V) oxides, e.g. α-Fe2O319 and V2O5.6 The total first-discharge capacity is 1693 mAh g−1. The subsequent charge reaction (Li+ deintercalation) is represented by the S-shaped curve and gives a capacity of 1232 mAh g−1 (release of 7.9 Li per unit formula of FeVO4). The irreversible capacity loss of 461 mAh g−1 (i.e., Δxirr = 2.9) can be attributed to the formation of the SEI film.20 The second discharge curve displays a similar shape to the first one suggesting that the charge–discharge process is highly reversible. The 2nd discharge and charge capacities are 1316 and 1215 mAh g−1, respectively, indicating a much reduced irreversible capacity loss of 101 mAh g−1. After 100 cycles, the electrode can still exhibit a reversible capacity of 1237 mAh g−1, corresponding to 94% of the 2nd discharge capacity (Fig. 2b). In addition, the Coulombic efficiency is 73% for the first cycle, which increases to close to 100% upon progressive cycling. The measured specific capacities of these amorphous FeVO4 nanosheets are comparable to that reported for the amorphous FeVO4 during the initial few cycles,1 which is higher than that of crystalline FeVO4.
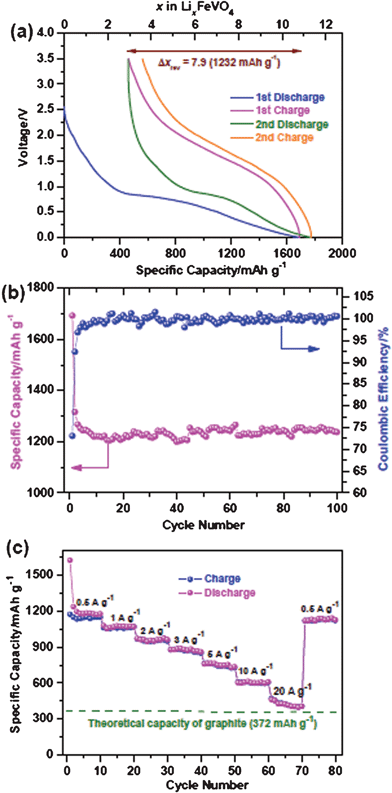 | ||
| Fig. 2 Anode performance of amorphous FeVO4 nanosheet arrays on a SS foil in the voltage range of 0.005–3.5 V (vs. Li/Li+). (a) Galvanostatic charge–discharge voltage profiles and (b) cycling performance at a current density of 200 mA g−1. (c) Charge–discharge capacities at various current densities of 0.5 to 20 A g−1. | ||
In order to clarify the electrochemical processes of amorphous FeVO4, additional measurements including cyclic voltammograms (CV) and ex situ XPS were also performed. Fig. S7 (ESI†) depicts representative CV plots of amorphous FeVO4 for the first three cycles in the voltage range of 3.5 to 0.005 V at a scan rate of 0.2 mV s−1. The peaks in the CV curves are distinct with two cathodic ones located at ∼1.4 V (weak) and ∼0.7 V (strong), and a broad anodic one at ∼1.7 V, which suggests solid solution behaviors and is consistent with the sloping voltage curves shown in Fig. 2a. The oxidation states of Fe and V elements on the fully lithiated and delithiated amorphous FeVO4 during the first cycle were examined by ex situ XPS technique (Fig. S8, ESI†). In the Fe 2p spectrum (Fig. S8a, ESI†), two peaks with the binding energies of 707.1 eV (Fe 2p3/2) and 720.0 eV (Fe 2p1/2) correspond to the Fe0 state,21 which indicates that the iron exists in the metallic form after the first discharge. The V 2p spectrum at the end of the first discharge is fitted to two components (Fig. S8b, ESI†). The smaller component is assigned to V3+ while the major component, appearing at lower binding energies, is assigned to V2+. The ratio of V2+:V3+ is estimated to be 32:68, which implies that the average oxidation state of V is 2.3. The Fe0 is oxidized to +3 again during the subsequent delithiation process at 3.5 V (Fig. S8c, ESI†). However, as shown in the XPS V 2p spectrum (Fig. S8d, ESI†), the vanadium is not fully recovered to +5, with the existence of V4+ (V4+:V5+ = 34:66), indicating an average V oxidation state of 4.7. These results are similar to previous XANES measurements performed on the fully lithiated and delithiated crystalline triclinic FeVO4.3 Thus, the discharge process corresponds to the full reduction of iron (Fe3+ → Fe0) and a partial reduction of vanadium (V5+ → V2.3+); and the iron and vanadium then change to +3.0 and +4.7 at the end of subsequent charge process, respectively.
Based on the above results, 5.4 Li per unit formula of FeVO4 can be reversibly stored. The calculated theoretical capacity of FeVO4 would be 847 mAh g−1, which, however, is not sufficient to explain the higher capacity of FeVO4 obtained experimentally, e.g., 1237 mAh g−1 during the 100th discharge. According to previous reports,2,3 the anion (i.e., oxygen) plays an important role during the lithium reaction, it can act as a redox center leading to possible Fe–“O–Li” and V–“O–Li” interactions. This can result in an enhancement in the specific capacity. The formation of Li–O interactions was also discovered during the lithium introduction in molybdenum oxides.22
Good high-C rate performances are desired in developing high power/fast charging lithium ion batteries. The cycling responses of the FeVO4 nanosheet array electrodes at different C rates were hence evaluated and are shown in Fig. 2c. The electrode shows discharge capacities of 1237, 1054, 961, 878, 764 and 601 mAh g−1 during the 2nd cycle at current densities of 0.5 (0.4 C), 1 (0.8 C), 2 (1.5 C), 3 (2.3 C), 5 (3.8 C) and 10 A g−1 (8 C), respectively. Even at a very high current density of 20 A g−1 (15 C), the FeVO4 electrode can still deliver a 2nd cycle discharge capacity of 453 mAh g−1, which is higher than the theoretical capacity of graphite (372 mAh g−1). Furthermore, the specific capacity could recover and be sustained at about 1120 mAh g−1 when the current density was returned to 0.5 A g−1. As compared to reported high-performance anodes,23–25 the above demonstrated high-rate Li storage performance of the FeVO4 electrode is excellent.
In summary, we have developed a facile template-free and catalyst-free CVD method to grow amorphous FeVO4 nanosheet arrays on SS foil. The resulting nanosheet arrays can be directly used as Li-ion battery anodes without addition of any ancillary materials. These FeVO4 2D arrays show excellent Li storage properties with high capacities, stable cyclability and excellent rate capability.
Acknowledgements
The authors gratefully acknowledge AcRF Tier 1 RG 31/08 of MOE (Singapore), NRF2009EWT-CERP001-026 (Singapore), the Singapore Ministry of Education (MOE2010-T2-1-017), A*STAR SERC grant 1021700144 and the Singapore MPA 23/04.15.03 RDP 009/10/102 and MPA 23/04.15.03 RDP 020/10/113 grants.References
- S. Denis, E. Baudrin, M. Touboul and J. M. Tarascon, J. Electrochem. Soc., 1997, 144, 4099–4109 CrossRef CAS.
- S. Denis, R. Dedryvere, E. Baudrin, S. Laruelle, M. Touboul, J. Olivier-Fourcade, J. C. Jumas and J. M. Tarascon, Chem. Mater., 2000, 12, 3733–3739 CrossRef CAS.
- S. Denis, E. Baudrin, F. Orsini, G. Ouvrard, M. Touboul and J. M. Tarascon, J. Power Sources, 1999, 81, 79–84 CrossRef.
- P. Poizot, E. Baudrin, S. Laruelle, L. Dupont, M. Touboul and J. M. Tarascon, Solid State Ionics, 2000, 138, 31–40 CrossRef CAS.
- D. W. Kim, Y. D. Ko, J. G. Park and B. K. Kim, Angew. Chem., Int. Ed., 2007, 46, 6654–6657 CrossRef CAS.
- X. H. Rui, J. X. Zhu, D. Sim, C. Xu, Y. Zeng, H. H. Hng, T. M. Lim and Q. Y. Yan, Nanoscale, 2011, 3, 4752–4758 RSC.
- X. H. Rui, J. X. Zhu, W. L. Liu, H. T. Tan, D. H. Sim, C. Xu, H. Zhang, J. Ma, H. H. Hng, T. M. Lim and Q. Y. Yan, RSC Adv., 2011, 1, 117–122 RSC.
- J. X. Zhu, Z. Y. Lu, M. O. Oo, H. H. Hng, J. Ma, H. Zhang and Q. Y. Yan, J. Mater. Chem., 2011, 21, 12770–12776 RSC.
- J. X. Zhu, Z. Y. Yin, H. Li, H. T. Tan, C. L. Chow, H. Zhang, H. H. Hng, J. Ma and Q. Y. Yan, Small, 2011, 7, 3458–3464 CrossRef CAS.
- Y. Q. Song, S. S. Qin, Y. W. Zhang, W. Q. Gao and J. P. Liu, J. Phys. Chem. C, 2010, 114, 21158–21164 CAS.
- J. P. Liu, Y. Y. Li, X. T. Huang, R. M. Ding, Y. Y. Hu, J. Jiang and L. Liao, J. Mater. Chem., 2009, 19, 1859–1864 RSC.
- Y. Q. Fan, H. B. Shao, J. M. Wang, L. A. Liu, J. Q. Zhang and C. N. Cao, Chem. Commun., 2011, 47, 3469–3471 RSC.
- Y. G. Li, B. Tan and Y. Y. Wu, Nano Lett., 2008, 8, 265–270 CrossRef CAS.
- S. Saadat, Y. Y. Tay, J. X. Zhu, P. F. Teh, S. Maleksaeedi, M. M. Shahjamali, M. Shakerzadeh, M. Srinivasan, B. Y. Tay, H. H. Hng, J. Ma and Q. Y. Yan, Chem. Mater., 2011, 23, 1032–1038 CrossRef CAS.
- R. Liu, J. Duay and S. B. Lee, Chem. Commun., 2011, 47, 1384–1404 RSC.
- C. K. Chan, H. L. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31–35 CrossRef CAS.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS.
- E. A. Olivetti, J. H. Kim, D. R. Sadoway, A. Asatekin and A. M. Mayes, Chem. Mater., 2006, 18, 2828–2833 CrossRef CAS.
- M. V. Reddy, T. Yu, C. H. Sow, Z. X. Shen, C. T. Lim, G. V. S. Rao and B. V. R. Chowdari, Adv. Funct. Mater., 2007, 17, 2792–2799 CrossRef CAS.
- X. H. Rui, N. Yesibolati and C. H. Chen, J. Power Sources, 2011, 196, 2279–2282 CrossRef CAS.
- J. Leveneur, G. I. N. Waterhouse, J. Kennedy, J. B. Metson and D. R. G. Mitchell, J. Phys. Chem. C, 2011, 115, 20978–20985 CAS.
- F. Leroux, G. R. Goward, W. P. Power and L. F. Nazar, Electrochem. Solid-State Lett., 1998, 1, 255–258 CrossRef CAS.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333–3338 CrossRef CAS.
- Y. Zhao, J. X. Li, Y. H. Ding and L. H. Guan, Chem. Commun., 2011, 47, 7416–7418 RSC.
- C. M. Ban, Z. C. Wu, D. T. Gillaspie, L. Chen, Y. F. Yan, J. L. Blackburn and A. C. Dillon, Adv. Mater., 2010, 22, E145–E149 CrossRef CAS.
Footnotes |
| † Electronic Supplementary Information (ESI) available: experimental details, scheme, temperature profile and supplementary XRD, FESEM, TEM, EDX, SAED, XPS. See DOI: 10.1039/c2ra20058a/ |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |