Electrocatalysis in MIRC reaction strategy: facile stereoselective approach to medicinally relevant spirocyclopropylbarbiturates from barbituric acids and activated olefins†
Evgeniya O.
Dorofeeva
*a,
Michail N.
Elinson
a,
Anatoly N.
Vereshchagin
a,
Nikita O.
Stepanov
a,
Ivan S.
Bushmarinov
b,
Pavel A.
Belyakov
a,
Olga O.
Sokolova
a and
Gennady I.
Nikishin
a
aN. D. Zelinsky Institute of Organic Chemistry, Leninsky pr. 47, Moscow, 119991, Russian Federation. E-mail: e.o.dorofeeva@gmail.com; Fax: +7(499)1355328; Tel: +7(499)1372944
bA. N. Nesmeyanov Institute of Organoelement Compounds, Vavilova str. 28, Moscow, 119991, Russian Federation. E-mail: ib@ineos.ac.ru; Fax: +7(499)1355085; Tel: +7(499)1359202
First published on 20th March 2012
Abstract
The combined electrolysis of barbituric acids and benzylidenemalononitriles or benzylidenecyanoacetates in methanol in an undivided cell in the presence of sodium bromide results in efficient MIRC (Michael-initiated ring-closure) formation of the corresponding spirocyclopropylbarbiturates in 45–93% yield. The electrocatalytic reaction proceeds smoothly under neutral and mild conditions with benzylidenemalononitriles or benzylidenecyanoacetates bearing both electron-donating and electron-withdrawing groups. NMR and single X-ray diffraction studies indicate that the electrocatalytic MIRC transformation of barbituric acids and benzylidenecyanoacetates results in the stereoselective formation of spirocyclopropanes with an (E)-configuration of aryl and alkoxycarboxylate substituents. The implication of electrocatalysis in the MIRC reaction strategy allows the combination of the synthetic virtues of both methods and accounts for an efficient approach to medicinally relevant spirocyclopropylbarbiturates avoiding inconvenient direct use of molecular halogen or halogenated substrates in accordance with the concepts of modern green chemistry.
Introduction
Pyrimidine-2,4,6-triones (barbiturates) belong to a widely-spread group of drugs used as sedatives, narcotics, anticonvulsants and antiepileptic agents.1 Currently, clinical applications of barbiturates are expanding in connection with their documented anti-invasive, antitumor and antiangiogenic efficiency.2 Spirobarbiturates have attracted special attention in the organic and pharmaceutical community due to the unique structural assembly and associated spectrum of biological properties.3 Thus, spirobarbiturates are known to exhibit neuropharmacological effects4 and are inhibitors of MMP-135 and dihydroorotate dehydrogenase (DHODase).6 Moreover, the corresponding 1-phenyl-5,7-diazaspiro[2.5]octane-4,6,8-trione was patented recently as a tumor necrosis factor-alpha (TNF-α) converting enzyme and matrix metalloproteinase inhibitor that could be potentially utilized in the treatment of various inflammatory, infectious, immunological or malignant diseases and conditions.7The conventional route to a barbituric cycle fused with a cyclopropyl ring implies a condensation of urea and 1,1-cyclopropyldicarboxylate esters in the presence of a base.6–8 Usual yields in such reactions are modest due to different side processes including ester hydrolysis, decarboxylation, transesterification and urea degradation. Moreover, requirements for dry solvents, high reaction temperatures and the application of strong bases dramatically limit the synthetic scope of this route.
Another approach to the spirocyclopropylbarbiturate framework utilizes direct cyclopropanation of barbituric acid derivatives. Thus, the reaction of carbenes or ylides with carbon–carbon double bonds of benzylidenebarbiturates,6,9 base promoted high-temperature alkylation of barbituric acid with dibromoethane10 and condensation of acetylenic esters and barbituric acid in the presence of triphenylarsine11 are known in the literature. Nevertheless, the most notable synthesis of substituted spirocyclopropylbarbiturates involves halogenated barbituric acids and refers to a Michael-initiated ring-closure (MIRC) reaction strategy, which has emerged as one of the most successful and convenient principles for the formation of a functionalized cyclopropane ring.12–14 The MIRC condensation of electron-deficient olefins either with activated dibromobarbiturate13 or N,N′-dialkylbarbituric acids in the presence of halogens14 afforded reasonable yields of the desired compounds. However, the first process was only realized in a single example, while the second reaction required up to 3 h to proceed. Moreover, both MIRC approaches required an inconvenient stoichiometric use of molecular halogen or halogenated substrates in the overall synthetic scheme and did not imply any stereoselectivity aspects.
Thus, each of the known methods for the synthesis of the 4,6,8-trioxo-5,7-diazaspiro[2.5]octane framework has its own merit but includes some obvious drawbacks ranging from a long reaction time to moderate yields, harsh reaction conditions and the stoichiometric application of molecular halogen or halogenated substrates. All these factors impact negatively on the practical use of the described processes and limit their application, especially when considering contemporary “Green Chemistry” demands. Taking into account the biomedical potential of spirocyclopropylbarbiturates as well as the discussed drawbacks of the known synthetic procedures, a benign and stereoselective MIRC approach to the corresponding 4,6,8-trioxo-5,7-diazaspiro[2.5]octane family that avoids direct halogenations is of great interest to the organic, industrial and pharmaceutical communities, but it is yet to be developed.
Advances in electrosynthesis during the last few decades have provided organic chemists with a new and versatile synthetic device with great promise.15 One of the most fascinating up-to-date organic electrochemistry methods is indirect electrocatalytic oxidation that takes place in the presence of mediators or mediatory systems. These mediators allow the use of undivided cells and generally simplify the conduction of the process. Among a variety of mediators, the redox system, halide anion/halogen, is one of the most suitable for selective organic syntheses and large-scale processes.16 The electrolysis in an undivided cell in the presence of an alcohol as solvent and alkali metal halides affords the simultaneous generation of catalytic quantities of a base (alkoxide anion) at the cathode and halogen at the anode, which then activate the chemical oxidative cycle in solution and return back to the electrodes as initial halide anion and alcohol upon completion of the cycle. The mild catalytic nature of the mediatory oxidative process enables low current concentrations of the base and halogen along with a neutral pH of the reaction solution, which are key factors for the selectivity and high product yields in many halogen-promoted oxidative transformations.
The implications of electrocatalysis in halogen-dependent MIRCs are highly promising as it allows the combination of the synthetic virtues of conventional MIRC strategy with the efficiency and ecological benefits of the facile electrocatalytic procedure. Considering our experience in the electrocatalytic synthesis of functionally rich cyclopropanes17 as well as the biomedical applications of spirocyclopropylbarbiturates given above, we were prompted to design a convenient electrocatalytic MIRC methodology for the one-pot synthesis of functionalized 4,6,8-trioxo-5,7-diazaspiro[2.5]octanes that includes stereoselectivity possibilities and avoids the direct use of molecular halogen or halogenated substrates.
Results and discussion
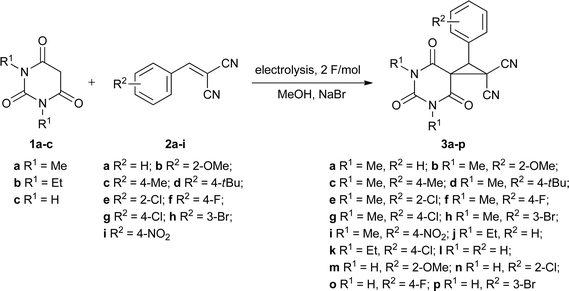
In the present study we report our results on the electrocatalytic MIRC reaction of barbituric acids and activated olefins (benzylidenemalononitriles or benzylidenecyanoacetates) under neutral and mild conditions by combined electrolysis in an undivided cell. The reaction is performed in methanol in the presence of sodium halides as mediators.First, to evaluate the synthetic potential of the proposed procedure, the electrocatalytic transformation of barbituric acid 1a and benzylidenemalononitrile 2a into 1-phenyl-5,7-diazaspiro[2.5]octane-4,6,8-trione 3a was studied (Scheme 1, Table 1).
 | ||
| Scheme 1 | ||
| Entry | Mediator | Electricity passed (F mol−1) | Current density (mA cm−2) | Temperature (°C) | Yield of 3a (%)b |
|---|---|---|---|---|---|
| a 1a (5 mmol), 2a (5 mmol), mediator (3 mmol), MeOH (20 mL), iron cathode (5 cm2), graphite anode (5 cm2). b Yield of isolated product. | |||||
| 1 | NaBr | 2 | 50 | 30 | 43 |
| 2 | NaBr | 2 | 100 | 30 | 65 |
| 3 | NaBr | 2 | 200 | 30 | 54 |
| 4 | NaBr | 2 | 100 | 20 | 73 |
| 5 | NaBr | 2 | 100 | 10 | 93 |
| 6 | NaBr | 2 | 100 | 0 | 55 |
| 7 | NaI | 2 | 100 | 10 | 57 |
The reaction protocol was optimized to achieve the highest yield of 3a with simultaneous minimization of the electrolysis time. Initially, excellent conversions (98–99%) of the starting compounds were obtained under 100 mA cm−2 and 200 mA cm−2 current densities after 2 F mol−1 of electricity had been passed (Table 1, entries 2 and 3). The current density 100 mA cm−2 (I = 500 mA, electrode surface 5 cm2) was found to be optimal for the electrochemically induced process. An increase of the current density up to 200 mA cm−2 (I = 1000 mA) resulted in a slight decrease in the reaction yield due to the possible activation of undesired direct electrochemical processes leading to oligomerization of the starting material and intermediate species.
A decrease in the electrolysis temperature has a dramatic impact on the yield of 3a (Table 1, entries 4–6). Thus, electrocatalysis of 1a and 2a under 100 mA cm−2 current density at 10 °C affords the corresponding spirocyclopropylbarbiturate 3a in the highest yield of 93% (Table 1, entry 5). A further decrease in the reaction temperature to 0 °C leads to the incomplete conversion of starting materials and only a 55% yield of the desired 4,6,8-trioxo-2-phenyl-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile 3a (Table 1, entry 6).
Sodium iodide is a less effective mediator compared to sodium bromide for the process studied. The electrolysis of barbituric acid 1a and benzylidenemalononitrile 2a in methanol at 10 °C in the presence of sodium iodide affords spirocyclopropylbarbiturate 3a in only a 57% yield (Table 1, entry 7). It is conceivable that the bromine formed at the anode during the electrolysis is a more effective oxidizer compared to iodine, and its presence promotes the chemical oxidative cycle in the solution.
Under the optimal conditions (sodium bromide as mediator, 100 mA cm−2 current density, I = 500 mA, 2 F mol−1 electricity passed (32 min reaction time), 10 °C reaction temperature) the electrolysis of barbituric acids 1a–c and benzylidenemalononitriles 2a–i in methanol in an undivided cell affords the corresponding 4,6,8-trioxo-5,7-diazaspiro[2.5]octanes 3a–p in 55–93% yields (Table 2). The developed electrocatalytic system offers a unique approach to the facile assembly of the spirocyclopropylbarbituric moiety. The reaction proceeds with benzylidenemalononitriles bearing both electron-donating and electron-withdrawing substituents in the phenyl ring. Moreover, products 3a–p are isolated by direct crystallization from the reaction mixture and do not require any further purification.
| Entry | Barbituric acid 1 | Olefin 2 | R 1 | R 2 | Product yield (%)b |
|---|---|---|---|---|---|
| a 1 (5 mmol), 2 (5 mmol), sodium bromide (3 mmol), methanol (20 mL), iron cathode (5 cm2), graphite anode (5 cm2), 10 °C, 100 mA cm−2 current density, 2 F mol−1 electricity passed. b Yield of isolated product. | |||||
| 1 | 1a | 2a | Me | H | 3a, 93 |
| 2 | 1a | 2b | Me | 2-OMe | 3b, 75 |
| 3 | 1a | 2c | Me | 4-Me | 3c, 77 |
| 4 | 1a | 2d | Me | 4-tBu | 3d, 78 |
| 5 | 1a | 2e | Me | 2-Cl | 3e, 61 |
| 6 | 1a | 2f | Me | 4-F | 3f, 68 |
| 7 | 1a | 2g | Me | 4-Cl | 3g, 75 |
| 8 | 1a | 2h | Me | 3-Br | 3h, 84 |
| 9 | 1a | 2i | Me | 4-NO2 | 3i, 74 |
| 10 | 1b | 2a | Et | H | 3j, 65 |
| 11 | 1b | 2g | Et | 4-Cl | 3k, 61 |
| 12 | 1c | 2a | H | H | 3l, 91 |
| 13 | 1c | 2b | H | 2-OMe | 3m, 55 |
| 14 | 1c | 2e | H | 2-Cl | 3n, 60 |
| 15 | 1c | 2f | H | 4-F | 3o, 65 |
| 16 | 1c | 2h | H | 3-Br | 3p, 68 |
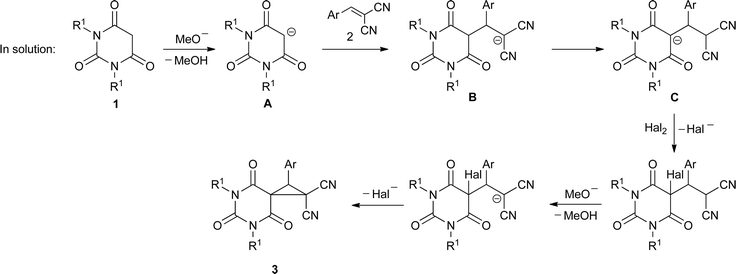
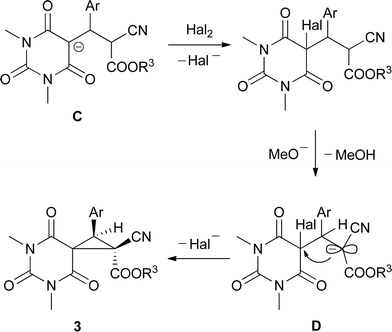
With the above results taken into consideration and the mechanistic data on the indirect electrochemical cyclopropanations previously performed by us,17 the following mechanism for the electrocatalytic MIRC transformation of barbituric acids 1 and benzylidenemalononitriles 2 into spirocyclopropylbarbiturates 3 is proposed.
Reactions at electrodes are usual for the applied mediatory system and lead to the formation of halogens at the anode and the deprotonation of methanol at the cathode (Scheme 2). The formation of either bromine or iodine at the anode is visually observed by the corresponding colour of the solution if electrolysis proceeds without stirring. The cathodic process results in the generation of methoxide anions and the liberation of hydrogen.
 | ||
| Scheme 2 | ||
The following processes represent a fairly complex pattern of equilibriums and cascade reactions in the solution. We assume that the major mechanistic route leading to spirocyclopropylbarbiturates 3 under the described electrocatalytic conditions could be outlined as follows (Scheme 3). First, deprotonation of barbituric acid 1 by a methoxide anion gives rise to the barbiturate anion A. Recently it was shown that the successful chemical synthesis of spirocyclopropylbarbiturates 3 in an alcoholic medium strictly requires the initial addition of sodium alkoxide to a mixture of barbituric acid and benzylidenemalononitrile giving rise to the intermediate Michael adduct, which is then selectively oxidized to 3 by sequentially adding bromine.14 Thus, the halogenation of A seems to be a minor process in our system, and Michael addition of the barbiturate anion to benzylidenemalononitrile 2 with the formation of adduct B determines the reaction pathway further. The adduct B should exist in equilibrium with adduct C by the proton migration possible under the conditions studied. Although both B and C could lead to the final spirocyclopropylbarbiturate 3 through the corresponding halogenation and further methoxide-promoted ring closure, a recent paper by Mayr's group suggests the dominant role of adduct C on the way to spirocyclopropylbarbiturates 3.18 Thus, Mayr et al. have shown that in aprotic solvents potassium salts of barbituric anions of type C were stable, did not give adducts of type B through proton migration and selectively reacted with methyl iodide on the 5-position of the pyrimidine ring. Moreover, upon protonation, anion C did not give its neutral form equal to that of anion B, but underwent a retro-Michael addition with the formation of barbituric acid and benzylidenemalononitrile (characteristic retro-Michael products for the unstable anion B) as well as intramolecular cyclization to the dihydropyrano[2,3-d]pyrimidine system, which we were also able to detect in our reaction mixtures when electricity as low as 0.1 F mol−1 was applied.19 All these data confirm an unambiguous and major involvement of the Michael adduct C in the reaction mechanism of the developed electrocatalytic process. Uptake of the adduct C by halogen followed by methoxide-assisted deprotonation and ring closure leads to spirocyclopropylbarbiturate 3.
 | ||
| Scheme 3 | ||
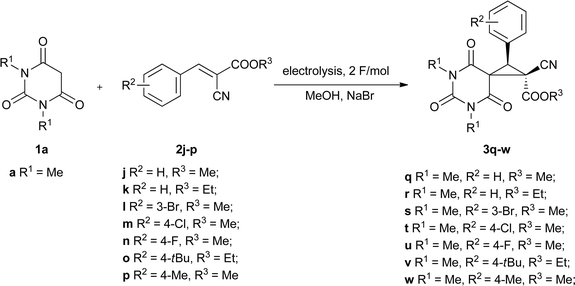
To widen the scope of the developed method and evaluate its stereoselectivity aspects, the electrocatalytic MIRC transformations of N,N′-dialkylbarbituric acid 1a and benzylidenecyanoacetates 2j–p were studied (Scheme 4). The electrocatalysis of 1a and methyl benzylidenecyanoacetate 2j using sodium bromide as the mediator under 100 mA cm−2 current density in methanol in an undivided cell at 10 °C reaction temperature afforded the corresponding 4,6,8-trioxo-5,7-diazaspiro[2.5]octane 3q in 42% yield (Table 3). The yield of 3q was also found to be dependent on the temperature of the process (Table 3, entries 1–4) giving the highest value of 57% when the electrolysis was carried out at 0 °C. Sodium iodide is a less efficient mediator for the process (Table 3, entry 5), similar to the electrocatalytic MIRC reaction of barbituric acids and benzylidenemalononitriles described above. Variation of the current density and the amount of electricity passed did not increase the yield of 3q.
 | ||
| Scheme 4 | ||
| Entry | Mediator | Electricity passed (F mol−1) | Time (min) | Temperature (°C) | Product yield (%)b |
|---|---|---|---|---|---|
| a 1a (5 mmol), 2j (5 mmol), mediator (3 mmol), MeOH (20 mL), iron cathode (5 cm2), graphite anode (5 cm2), current density 100 mA cm−2. b Yield of isolated product. | |||||
| 1 | NaBr | 2 | 32 | 20 | 40 |
| 2 | NaBr | 2 | 32 | 10 | 42 |
| 3 | NaBr | 2 | 32 | 0 | 57 |
| 4 | NaBr | 2 | 32 | −10 | 37 |
| 5 | NaI | 2 | 32 | 0 | 50 |
Under the optimal conditions found (sodium bromide as mediator, 100 mA cm−2 current density, I = 500 mA, 2 F mol−1 electricity passed (32 min reaction time), 0 °C reaction temperature) the electrolysis of barbituric acid 1a and benzylidenecyanoacetates 2j–p in methanol in an undivided cell affords the corresponding 4,6,8-trioxo-5,7-diazaspiro[2.5]octanes 3q–w in 45–71% yields (Table 4).
| Entry | Barbituric acid 1 | Olefin 2 | R 1 | R 2 | R 3 | Product yield (%)b |
|---|---|---|---|---|---|---|
| a 1 (5 mmol), 2 (5 mmol), sodium bromide (3 mmol), methanol (20 mL), iron cathode (5 cm2), graphite anode (5 cm2), 0 °C, current density 100 mA cm−2, 2 F mol−1 electricity passed. b Yield of isolated product. | ||||||
| 1 | 1a | 2j | Me | Me | H | 3q, 57 |
| 2 | 1a | 2k | Me | Et | H | 3r, 59 |
| 3 | 1a | 2l | Me | Me | 3-Br | 3s, 67 |
| 4 | 1a | 2m | Me | Me | 4-Cl | 3t, 57 |
| 5 | 1a | 2n | Me | Me | 4-F | 3u, 71 |
| 6 | 1a | 2o | Me | Et | 4-tBu | 3v, 47 |
| 7 | 1a | 2p | Me | Me | 4-Me | 3w, 45 |
It should be mentioned that the obtained spirocyclopropylbarbiturates 3q–w could exist as pairs of diastereoisomers with (E)- or (Z)-configuration of the aryl and carbomethoxy substituents relative to the cyclopropane ring. However, in the NMR spectra of 3q–w, only a single set of signals was identified, assuming the stereoselective formation of individual diastereoisomers in the developed electrocatalytic process. The structure of the spirocyclopropylbarbiturate 3q was further confirmed by a single-crystal X-ray diffraction study (Fig. 1). The X-ray diffraction data unambiguously support the (E)-configuration for 3q, with the phenyl and carbomethoxy substituents lying on different sides of the cyclopropane plane. Considering the facts given above, compounds 3q–w should also possess (E)-configuration.
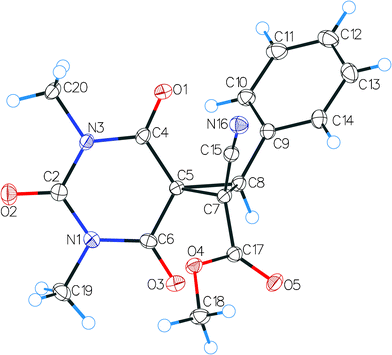 | ||
| Fig. 1 The overview of the 3q molecule by single-crystal X-ray diffraction. Atoms are represented by thermal displacement ellipsoids (p = 50%). | ||
The stereoselectivity of the developed electrocatalytic MIRC transformation of N,N′-dimethylbarbituric acid 1a and benzylidenecyanoacetates 2j–p could be explained by the following considerations. All the main mechanistic steps for the formation of spirocyclopropylbarbiturates 3q–w are equal to those proposed for the formation of spirocyclopropylbarbiturates 3a–p, including the dominant formation of the Michael adduct of type C with its following halogenation at the 5-position of the pyrimidine ring (Scheme 3). Further methoxide-promoted deprotonation gives rise to the conjugated anion D and the steric hindrance between the aryl and carboalkoxy substituents seems to be the main reason for the following stereoselective cyclization leading to a cyclopropane ring with (E)-disposition of the corresponding substituents (Scheme 5).
 | ||
| Scheme 5 | ||
Conclusions
In the course of our study we have developed a facile electrocatalytic MIRC approach to substituted spirocyclopropylbarbiturates using reasonable starting materials and avoiding the inconvenient stoichiometric use of molecular halogen or halogenated substrates. Thus, combined electrolysis of barbituric acids and benzylidenemalononitriles or benzylidenecyanoacetates in methanol in an undivided cell results in the formation of the corresponding spirocyclopropylbarbiturates in 45–93% yield. Notably, the developed electrocatalytic MIRC process includes a stereoselective feature and allows for the synthesis of the corresponding functionally rich spirocyclopropylbarbiturate with (E)-configuration of the aryl and alkoxycarboxylate substituents relative to the cyclopropane plane. The electrocatalytic reaction proceeds smoothly with benzylidenemalononitriles or benzylidenecyanoacetates bearing both electron-donating and electron-withdrawing groups. The mild chemical nature of mediatory electrolysis allows good substance yields and current efficiency along with superior stereoselectivity. The implication of electrocatalysis in the MIRC reaction strategy combines the synthetic virtues of both methods and represent a prominent procedure for the preparation of medicinally privileged spirocyclopropylbarbiturates with promising applications. Finally, the developed procedure utilizes simple equipment and an undivided cell, and is valuable from the viewpoint of an environmentally benign synthesis and large-scale processes.Experimental section
General remarks
All melting points were measured with a Gallenkamp melting point apparatus and were uncorrected. 1H and 13C NMR spectra were recorded with Bruker AM-300 and Bruker Avance II 300 spectrometers at ambient temperature in DMSO-d6 solutions using Me4Si as the internal standard. IR spectra were registered with a SPECORD M82 spectrometer in KBr pellets. Mass-spectra (EI = 70 eV) were obtained directly with a Finningan MAT INCOS 50 spectrometer. Methanol with less than 1% water content was used without additional purification.Typical electrolysis procedure
A solution of barbituric acid (5 mmol), benzylidenemalononitrile or benzylidenecyanoacetate (5 mmol) and sodium bromide (0.31 g, 3 mmol) in 20 mL of methanol was electrolyzed in an undivided cell equipped with a magnetic stirrer, a graphite anode and an iron cathode under a constant current density of 100 mA cm−2 (I = 500 mA, electrodes square 5 cm2) either at 10 °C or at 0 °C, depending on the substrate, until 2 F mol−1 of electricity was passed (process time 32 min). After the electrolysis was finished, the reaction mixture was gently concentrated to one fifth of the initial volume (ca. 4 mL) to crystallize the solid product, which was then filtered off, rinsed with ice-cold methanol (2 × 2 mL), and dried under reduced pressure. Products 3a–p did not require any further purification. In the case of compounds 3q–w, an additional recrystallization from ethyl acetate was performed.Single crystals of C17H15N3O53q were grown from an acetone–hexane (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture. The X-ray diffraction (XRD) measurements were performed on a Bruker APEX II diffractometer. The crystal was kept at 100 K during data collection. Using Olex2,20 the structure was solved with the XS structure solution program using Direct Methods and refined with the XL21 refinement package using Least Squares minimization.
:1) mixture. The X-ray diffraction (XRD) measurements were performed on a Bruker APEX II diffractometer. The crystal was kept at 100 K during data collection. Using Olex2,20 the structure was solved with the XS structure solution program using Direct Methods and refined with the XL21 refinement package using Least Squares minimization.
Crystal data
C17H15N3O5, M = 341.32, orthorhombic, a = 9.4235(11) Å, b = 10.3957(12) Å, c = 15.9888(18) Å, V = 1566.3(3) Å3, T = 100, space group P212121 (no. 19), Z = 4, μ(Mo-Kα) = 0.109, 20970 reflections measured, 2756 unique (Rint = 0.0365), were used in all calculations. The final wR2 was 0.0852 (all data) and R1 was 0.0319 (> 2 sigma(I)). The crystallographic data reported for 3q are available in the ESI.†
5,7-Dimethyl-4,6,8-trioxo-2-phenyl-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3a)
White solid. Yield 1.43 g (93%). M.p. 259–260 °C (lit.,14 259–260 °C). δH (300 MHz, DMSO-d6) 3.12 (s, 3 H, CH3), 3.27 (s, 3 H, CH3), 4.36 (s, 1 H, CH), 7.32–7.40 (m, 3 H, Ar), 7.44–7.50 (m, 2 H, Ar).2-(2-Methoxyphenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3b)
Yellowish solid. Yield 1.28 g (75%). M.p. 229–230 °C (lit.,14 229–230 °C). δH (300 MHz, DMSO-d6) 3.14 (s, 3 H, CH3), 3.30 (s, 3 H, CH3), 3.72 (s, 3 H, CH3), 4.00 (s, 1 H, CH), 6.96–7.08 (m, 2 H, Ar), 7.34–7.39 (m, 2 H, Ar).5,7-Dimethyl-2-(4-methylphenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3c)
White solid. Yield 1.24 g (77%). M.p. 203–204 °C. δH (300 MHz, DMSO-d6): 2.30 (s, 3 H, CH3), 3.12 (s, 3 H, CH3), 3.26 (s, 3 H, CH3), 4.29 (s, 1 H, CH), 7.17 (d, J = 7.9 Hz, 2 H, Ar), 7.33 (d, J = 7.9 Hz, 2 H, Ar). δC (75 MHz, DMSO-d6) 20.7, 23.4, 28.5, 29.0, 41.3, 43.9, 110.8, 112.4, 125.5, 128.7 (2 C), 129.0 (2 C), 137.6, 150.8, 160.4, 162.9. νmax/cm−1 2252, 1692, 1688, 1684, 1444, 1384, 1300, 752 cm−1. MS (EI, 70 eV): m/z (%) = 322 (16) [M+], 295 (23), 265 (18), 208 (74), 180 (100), 165 (52), 153 (35), 140 (25), 115 (31), 91 (28). C17H14N4O3 (322.22): calcd. C 63.35, H 4.38, N 17.38; found (%): C 63.28, H 4.43, N 17.23.2-(4-tert-Butylphenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3d)
White solid. Yield 1.42 g (78%). M.p. 218–219 °C. δH (300 MHz, DMSO-d6) 1.28 (s, 9 H, CH3), 3.12 (s, 3 H, CH3), 3.26 (s, 3 H, CH3), 4.28 (s, 1 H, CH), 7.17 (d, J = 7.8 Hz, 2 H, Ph), 7.40 (d, J = 7.8 Hz, 2 H, Ph). δC (75 MHz, DMSO-d6) 23.5, 28.7, 29.2, 31.1 (3 C), 34.4, 41.3, 43.9, 111.0, 112.5, 125.1 (2 C), 125.5, 128.9 (2 C), 150.8, 150.9, 160.6, 163.0. νmax/cm−1 2252, 1708, 1692, 1684, 1444, 1396, 1384, 752. MS: m/z (%) = 364 (18) [M+], 350 (15), 349 (70), 299 (14) 292 (37), 235 (12), 207 (21), 194 (10), 140 (12), 57 (100). C20H20N4O3 (364.40): calcd. C 65.92, H 5.53, N 15.38; found C 65.83, H 5.67, N 15.28.2-(2-Chlorophenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3e)
White solid. Yield 1.05 g (61%). M.p. 255–256 °C. δH (300 MHz, DMSO-d6) 3.15 (s, 3 H, CH3), 3.38 (s, 3 H, CH3), 4.32 (s, 1 H, CH), 7.36–7.45 (m, 2 H, Ar), 7.52–7.58 (m, 2 H, Ar). δC (75 MHz, DMSO-d6) 24.4, 28.6, 29.1, 41.2, 42.4, 110.4, 111.9, 126.3, 126.9, 129.2, 130.3, 131.5, 133.4, 150.5, 160.4, 162.5. MS: m/z (%) = 344 (1) [37C1, M+], 342 (3) [35C1, M+], 307 (100), 250 (9), 228 (15), 193 (24), 165 (40), 138 (10), 75 (14), 56 (23). νmax/cm−1 2252, 1700, 1680, 1456, 1424, 1384, 1300, 756. C16H11ClN4O3 (342.74): calcd. C 56.07, H 3.23, Cl 10.34, N 16.35; found C 55.99, H 3.31, Cl 10.28, N 16.28.2-(4-Fluorophenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3f)
White solid. Yield 1.11 g (68%). M.p. 221–222 °C. δH (300 MHz, DMSO-d6) 3.12 (s, 3 H, CH3), 3.27 (s, 3 H, CH3), 4.32 (s, 1 H, CH), 7.21 (m, 2 H, Ph), 7.53 (dd, J1 = 8.8 Hz, J2 = 5.6 Hz, 2 H, Ph). δC (75 MHz, DMSO-d6) 23.7, 28.5, 29.0, 41.3, 43.0, 110.7, 112.2, 115.0 (d, J = 21.7 Hz, 2 C), 124.8 (d, J = 2.8 Hz), 131.5 (d, J = 8.3 Hz, 2 C), 150.8, 160.5, 161.9 (d, J = 245.3 Hz), 162.8. νmax/cm−1 3436, 3029, 2254, 1750, 1685, 1518, 1457, 1382, 1295, 753. MS: m/z (%) = 327 (11) [M+], 326 (24) [M+], 301 (8), 300 (11), 263 (24), 262 (48), 212 (100), 184 (98), 101 (42), 83 (32). C16H11FN4O3 (326.28): calcd. C 58.90, H 3.40, F 5.82, N 17.17; found (%): C 58.81, H 3.46, F 5.73, N 17.03.2-(4-Chlorophenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3g)
White solid. Yield 1.29 g (75%). M.p. 206–208 °C. δH (300 MHz, DMSO-d6) 3.14 (s, 3 H, CH3), 3.26 (s, 3 H, CH3), 4.35 (s, 1 H, CH), 7.44 (d, J = 8.80 Hz, 2 H, Ar), 7.51 (d, J = 8.80 Hz, 2 H, Ar). δC (75 MHz, DMSO-d6) 23.6, 28.5, 28.9, 41.2, 42.8, 110.6, 112.1, 127.7, 128.1 (2 C), 131.1 (2 C), 133.0, 150.8, 160.5, 162.7. νmax/cm−1 3436, 2252, 1684, 1458, 1426, 1386, 1300, 1091, 1015, 753 cm−1. MS: m/z (%) = 343 (3) [M+], 342 (35) [M+], 316 (17), 280 (38), 278 (88), 229 (65), 203 (26), 201 (24), 166 (100), 136 (41). C16H11ClN4O3 (342.74): calcd. C 56.07, H 3.23, Cl 10.34, N 16.35; found C 55.98, H 3.29, Cl 10.29, N 16.30.2-(3-Bromophenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3h)
Yellowish solid. Yield 1.63 g (84%). M.p. 220–222 °C (lit.,14 217–219 °C). δH (300 MHz, DMSO-d6) 3.12 (s, 3 H, CH3), 3.28 (s, 3 H, CH3), 4.39 (s, 1 H, CH), 7.35 (t, J = 7.9 Hz, 1 H, Ph), 7.50–7.57 (m, 2 H, Ph), 7.77–7.79 (m, 1 H, Ph).5,7-Dimethyl-2-(4-nitrophenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3i)
Yellowish solid. Yield 1.31 g (74%). M.p. 219–221 °C. δH (300 MHz, DMSO-d6) 3.12 (s, 3 H, CH3), 3.30 (s, 3 H, CH3), 4.55 (s, 1 H, CH), 7.82 (d, J = 8.4 Hz, 2 H, Ar), 8.24 (d, J = 8.4 Hz, 2 H, Ar). δC (75 MHz, DMSO-d6) 23.7, 28.5, 29.0, 41.4, 42.3, 110.5, 111.9, 123.0 (2 C), 130.8 (2 C), 136.4, 147.2, 150.8, 160.6, 162.5. νmax/cm−1 2256, 1700, 1680, 1520, 1460, 1424, 1388, 1348, 756. MS: m/z (%) = 353 (26) [M+], 326 (8), 296 (16), 239 (100), 211 (32), 194 (22), 165 (72), 138 (34), 117 (29), 75 (29). C16H11N5O5 (353.29): calcd. C 54.39, H 3.14, N 19.82; found C 54.23, H 3.22, N 19.78.5,7-Diethyl-4,6,8-trioxo-2-phenyl-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3j)
White solid. Yield 1.10 g (65%). M.p. 186–187 °C (lit.,14 186–188 °C). δH (300 MHz, DMSO-d6) 1.18 (t, J = 7.0 Hz, 3 H, CH3), 1.33 (t, J = 7.0 Hz, 3 H, CH3), 3.90–4.00 (m, 2 H, CH2), 4.10 (q, J = 7.0 Hz, 2 H, CH2), 4.29 (s, 1 H, CH), 7.22–7.32 (m, 2 H, Ar), 7.36–7.46 (m, 3 H, Ar).2-(4-Chlorophenyl)-5,7-diethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3k)
White solid. Yield 1.13 g (61%). M.p. 209–210 °C. δH (300 MHz, DMSO-d6) 1.19 (t, J = 7.1 Hz, 3 H, CH3), 1.32 (t, J = 7.1 Hz, 3 H, CH3), 3.96 (q, J = 7.0 Hz, 2 H, CH2), 4.05–4.15 (m, 2 H, CH2), 4.25 (s, 1 H, CH), 7.25 (d, J = 7.3 Hz, 2 H, Ar), 7.41 (d, J = 7.3 Hz, 2 H, Ar). δC (75 MHz, DMSO-d6) 12.54, 12.64, 23.70, 36.99, 37.54, 41.35, 42.74, 110.74, 112.24, 127.85 (2 C), 128.06 (2 C), 131.25, 133.05, 149.93, 160.20, 162.37. νmax/cm−1 2252, 1704, 1684, 1448, 1416, 1352, 1316, 760. MS: m/z (%) = 370 (2) [M+], 335 (5), 306 (3), 228 (15), 200 (20), 165 (87), 138 (14), 70 (100), 56 (89). C18H15ClN4O3 (370.79): calcd. C 58.31, H 4.08, Cl 9.56, N 15.11; found C 58.19, H 4.16, Cl 9.51, N 15.20.4,6,8-Trioxo-2-phenyl-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3l)
White solid. Yield 1.27 g (91%). M.p. 274–276 °C. δH (300 MHz, DMSO-d6) 4.23 (s, 1 H, CH), 7.34–7.46 (m, 5 H, Ph), 11.66 (s, 1 H, NH), 11.88 (s, 1 H, NH). δC (75 MHz, DMSO-d6) 22.5, 40.6, 43.1, 111.0, 112.5, 128.2 (2 C), 128.3, 128.7, 129.4 (2 C), 150.2, 161.8, 164.3. νmax/cm−1 3264, 2264, 1764, 1736, 1708, 1400, 1364, 1284, 1200, 768. MS: m/z (%) = 381 (7) [M+], 280 (19) [M+], 254 (11), 217 (98), 215 (66), 195 (52), 173 (40), 167 (78), 165 (26), 155 (100). C14H8N4O3 (280.24): calcd. C 60.00, H 2.88, N 19.99; found C 59.92, H 3.01, N 19.91.2-(2-Methoxyphenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3m)
White solid. Yield 0.85 g (55%). M.p. 269 °C. δH (300 MHz, DMSO-d6) 3.72 (s, 3 H, CH3), 3.86 (s, 1 H, CH), 6.95–7.05 (m, 2 H, Ph), 7.33–7.40 (m, 2 H, Ph), 11.83 (br s, 2 H, NH). δC (75 MHz, DMSO-d6) 22.3, 40.1, 40.4, 55.6, 110.9, 112.5 (2 C), 116.4, 120.3, 130.0, 130.1, 150.1, 156.9, 161.8, 164.6. νmax/cm−1 3365, 3095, 2249, 1729, 1706, 1600, 1418, 1292, 1018, 755. MS: m/z (%) 311 (12) [M+], 310 (50) [M+], 245 (48), 224 (100), 215 (63), 181 (86), 172 (50), 119 (54), 91 (74), 77 (90). C15H10N4O4 (310.26): C 58.07, H 3.25, N 18.06; found C 57.98, H 3.32, N 18.02.2-(2-Chlorophenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3n)
White solid. Yield 0.95 g (60%). M.p. 266–267 °C. δH (300 MHz, DMSO-d6) 4.17 (s, 1 H, CH), 7.36–7.44 (m, 2 H, Ph), 7.50–7.62 (m, 2 H, Ph), 11.84 (s, 1 H, NH), 12.08 (s, 1 H, NH). δC (75 MHz, DMSO-d6) 23.2, 40.7, 41.7, 110.7, 112.2, 126.8, 127.1, 129.3, 130.5, 131.5, 133.6, 150.1, 161.9, 164.1. νmax/cm−1 3253, 3130, 2254, 1768, 1737, 1712, 1428, 1400, 1363, 763. MS: m/z (%) 316 (3) [M+], 314 (7) [M+], 279 (30), 254 (4), 215 (100), 203 (14), 193 (21), 172 (53), 66 (28), 43 (55). C14H7ClN4O3 (314.68): calcd. C 53.43, H 2.24, Cl 11.27, N 17.80; found C 53.34, H 2.32, Cl 11.20, N 17.84.2-(4-Fluorophenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3o)
White solid. Yield 0.97 g (65%). M.p. 264 °C. δH (300 MHz, DMSO-d6) 4.17 (s, 1 H, CH), 7.16–7.22 (m, 2 H, Ph), 7.50–7.54 (m, 2 H, Ph), 11.72 (br s, 2 H, NH). δC (75 MHz, DMSO-d6) 22.8, 40.7, 42.2, 110.9, 112.4, 115.0 (d, J = 21.8 Hz, 2 C); 125.0 (d, J = 2.9 Hz), 131.7 (d, J = 8.3 Hz, 2 C), 150.3, 161.9, 162.0 (d, J = 244,8 Hz), 164.2. νmax/cm−1 3272, 3007, 2858, 2254, 1720, 1513, 1421, 1354, 861, 785. MS: m/z (%) = 298 (7) [M+], 271 (24), 233 (42), 212 (16), 184 (33), 172 (100), 145 (37), 123 (39), 66 (32), 43 (40). C14H7FN4O3 (298.23): calcd. C 56.38, H 2.37, F 6.37, N 18.79; found C 56.30, H 2.42, F 6.33, N 18.73.2-(3-Bromophenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1,1-dicarbonitrile (3p)
White solid. Yield 1.22 g (68%). M.p. 252 °C. δH (300 MHz, DMSO-d6) 4.24 (s, 1 H, CH), 7.33 (t, J = 7.8 Hz, 1 H, Ph), 7.51–7.54 (m, 2 H, Ph), 7.80 (s, 1 H, Ph), 12.70 (br s, 2 H, NH). δC (75 MHz, DMSO-d6) 22.7, 40.6, 41.6, 110.8, 112.3, 121.2, 128.5, 130.3, 131.2, 131.4, 132.3, 150.3, 162.0, 164.1. νmax/cm−1 3338, 3298, 3017, 2822, 2253, 1721, 1596, 1412, 1333, 755. MS: m/z (%) 360 (4) [M+], 358 (4) [M+], 295 (23), 293 (23), 234 (60), 232 (60), 153 (100), 126 (30), 101 (25), 43 (70). C14H7BrN4O3 (359.13): calcd. C 46.82, H 1.96, Br 22.25, N 15.60; found C 46.78, H 2.07, Br 22.20, N 15.57.Methyl (1R*,2S*)-1-cyano-5,7-dimethyl-4,6,8-trioxo-2-phenyl-5,7-diazaspiro[2.5]octane-1-carboxylate (3q)
White solid. Yield 0.98 g (57%). M.p. 204 °C. δH (300 MHz, DMSO-d6) 1.25 (t, J = 7.0 Hz, 3 H, CH3), 3.09 (s, 3 H, CH3), 3.20 (s, 3 H, CH3), 3.83 (s, 3 H, CH3), 4.04 (s, 1 H, CH), 7.30–7.45 (m, 5 H, Ar). δC (75 MHz, DMSO-d6) 28.5, 28.9, 36.4, 41.1, 42.7, 54.3, 112.5, 128.2, 128.3 (2 C), 128.9 (2 C), 129.4, 150.6, 160.8, 162.2, 164.3. νmax/cm−1 3016, 2960, 2244, 1752, 1692, 1676, 1428, 1300, 1096, 752. MS: m/z (%) 342 (1) [M+], 341 (12) [M+], 283 (60), 282 (100), 244 (9), 197 (24), 168 (35), 156 (21), 140 (63), 57 (48). C17H15N3O5 (341.32): calcd. C 59.82, H 4.43, N 12.31; found C 59.75, H 4.50, N 12.25.Ethyl (1R*,2S*)-1-cyano-5,7-dimethyl-4,6,8-trioxo-2-phenyl-5,7-diazaspiro[2.5]octane-1-carboxylate (3r)
White solid. Yield 1.04 g (59%). M.p. 187 °C. δH (300 MHz, DMSO-d6) 1.25 (t, J = 7.0 Hz, 3 H, CH3), 3.09 (s, 3 H, CH3), 3.20 (s, 3 H, CH3), 4.04 (s, 1 H, CH), 4.29 (q, J = 7.0 Hz, 2 H, CH2), 7.38–7.52 (m, 5 H, Ar). δC (75 MHz, DMSO-d6) 13.6, 28.5, 28.9, 36.7, 41.0, 42.4, 63.5, 112.6, 128.3 (2 C), 128.7 (2 C), 128.9, 129.4, 150.7, 160.8, 161.6, 164.1. νmax/cm−1 3272, 3007, 2858, 2254, 1720, 1513, 1421, 1354, 861, 785. MS: m/z (%) 356 (2) [M+], 355 (11) [M+], 328 (2), 283 (72), 282 (100), 244 (9), 168 (69), 141 (59), 140 (99), 128 (18). C18H17N3O5 (355.34): calcd. C 60.84, H 4.83, N 11.83; found C 60.74, H 4.90, N 11.90.Methyl (1R*,2S*)-2-(3-bromophenyl)-1-cyano-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1-carboxylate (3s)
White solid. Yield 1.41 g (67%). M.p. 218 °C. δH (300 MHz, DMSO-d6) 3.11 (s, 3 H, CH3), 3.20 (s, 3 H, CH3), 3.83 (s, 3 H, CH3), 4.07 (s, 1 H, CH), 7.36 (t, J = 7.6 Hz, 1 H, Ar), 7.45 (d, J = 7.6 Hz, 1H, Ar), 7.56 (d, J = 7.6 Hz, 1 H, Ar), 7.68 (s, 1 H, Ar). δC (75 MHz, DMSO-d6) 28.5, 28.9, 36.4, 41.0, 41.2, 54.4, 112.3, 121.4, 127.9, 130.4, 131.1, 131.8, 132.1, 150.7, 160.9, 161.9, 164.0. νmax/cm−1 3008, 2960, 2244, 1756, 1684, 1432, 1288, 1140, 1088, 780, 752. MS: m/z (%) 421 (19) [M+], 419 (21) [M+], 362 (100), 361 (28), 360 (98), 324 (5), 322 (7), 220 (27), 167 (26), 71 (30). C17H14BrN3O5 (419.21): calcd. C 48.59, H 3.36, Br 19.02, N 10.00; found C 48.48, H 3.42, Br 19.15, N 9.93.Methyl (1R*,2S*)-2-(4-chlorophenyl)-1-cyano-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1-carboxylate (3t)
White solid. Yield 1.03 g (57%). M.p. 195 °C. δH (300 MHz, DMSO-d6) 3.09 (s, 3 H, CH3), 3.19 (s, 3 H, CH3), 3.83 (s, 3 H, CH3), 4.04 (s, 1 H, CH), 7.35–7.55 (m, 4 H, Ar). δC (75 MHz, DMSO-d6) 22.5, 22.8, 36.5, 41.1, 41.7, 54.4, 112.4, 128.4 (2 C), 128.5, 130.9 (2 C), 133.1, 150.7, 160.8, 162.0, 164.1. νmax/cm−1 3008, 2960, 2244, 1756, 1684, 1428, 1232, 1088, 1020, 752. MS: m/z (%) 377 (4) [M+], 375 (11) [M+], 318 (54), 316 (100), 231 (8), 204 (19), 202 (27), 174 (43), 135 (21), 75 (22). C17H14ClN3O5 (375.76): calcd. C 54.34, H 3.76, Cl 9.43, N 11.18; found C 54.40, H 3.70, Cl 9.37, N 11.26.Methyl (1R*,2S*)-1-cyano-2-(4-fluorophenyl)-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1-carboxylate (3u)
White solid. Yield 1.28 g (71%). M.p. 217 °C. δH (300 MHz, DMSO-d6) 3.09 (s, 3 H, CH3), 3.19 (s, 3 H, CH3), 3.83 (s, 3 H, CH3), 4.02 (s, 1 H, CH), 7.20–7.26 (m, 2 H, Ar), 7.45–7.49 (m, 2 H, Ar). δC (75 MHz, DMSO-d6) 28.5, 28.9, 36.6, 41.1, 41.8, 54.4, 112.4, 115.2 (d, J = 21.7 Hz, 2 C); 125.6 (d, J = 2.8 Hz), 131.1 (d, J = 8.4 Hz, 2 C), 150.7, 160.8, 161.9 (d, J = 244,9 Hz), 162.1 164.2. νmax/cm−1 3008, 2968, 2248, 1756, 1680, 1516, 1428, 1216, 1096, 752. MS: m/z (%) 360 (6) [M+], 359 (15) [M+], 301 (32), 300 (100), 215 (8), 187 (15), 186 (26), 158 (72), 95 (26), 69 (23). C17H14ClN3O5 (359.31): calcd. C 56.83, H 3.93, F 5.29, N 11.69 found C 56.70, H 4.05, F 5,20, N 11.60.Ethyl (1R*,2S*)-2-(4-tert-butylphenyl)-1-cyano-5,7-dimethyl-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1-carboxylate (3v)
White solid. Yield 0.97 g (47%). M.p. 264 °C. δH (300 MHz, DMSO-d6) 1.20–1.29 (m, 12 H, CH3), 3.11 (s, 3 H, CH3), 3.20 (s, 3 H, CH3), 3.98 (s, 1 H, CH), 4.28 (q, J = 6.8 Hz, 2 H, CH2), 7.32 (d, J = 8.3 Hz, 2 H, Ar), 7.40 (d, J = 8.3 Hz, 2 H, Ar). δC (75 MHz, DMSO-d6) 13.6, 28.6, 28.9, 31.1 (3 C), 34.4, 36.9, 41.0, 42.4, 63.5, 112.8, 125.2 (2 C), 126.4, 128.7 (2 C), 150.6, 150.7, 160.9, 161.6, 164.2. νmax/cm−1 2964, 2904, 2224, 1752, 1688, 1456, 1376, 1264, 1092, 756. MS: m/z (%) 412 (3) [M+], 411 (19) [M+], 339 (9), 338 (100), 300 (11), 285 (21), 242 (26), 181 (10), 84 (21), 55 (58). C22H25N3O5 (411.45): calcd. C 64.22, H 6.12, N 10.21; found C 64.31, H 5.98, N 10.35.Methyl (1R*,2S*)-1-cyano-5,7-dimethyl-2-(4-methylphenyl)-4,6,8-trioxo-5,7-diazaspiro[2.5]octane-1-carboxylate (3w)
White solid. Yield 0.80 g (45%). M.p. 189 °C. δH (300 MHz, DMSO-d6) 2.30 (s, 3 H, CH3), 3.09 (s, 3 H, CH3), 3.19 (s, 3 H, CH3), 3.83 (s, 3 H, CH3), 3.98 (s, 1 H, CH), 7.18 (d, J = 7.9 Hz, 2 H, Ar), 7.28 (d, J = 7.9 Hz, 2 H, Ar). δC (75 MHz, DMSO-d6) 20.7, 28.5, 28.9, 36.4, 41.2, 42.8, 54.3, 112.6, 126.3, 128.8 (2 C), 128.9 (2 C), 137.6, 150.7, 160.8, 162.3, 164.3. νmax/cm−1 3023, 2963, 2245, 1754, 1687, 1450, 1374, 1228, 1088, 751. MS: m/z (%) 356 (3) [M+], 355 (14) [M+], 297 (54), 296 (84), 182 (77), 154 (92), 127 (49), 92 (62), 91 (100), 55 (32). C18H17N3O5 (355.34): calcd. C 60.84, H 4.82, N 11.83; found C 60.70, H 4.93, N 11.74.Acknowledgements
The authors gratefully acknowledge the financial support from the Grant of the President of the Russian Federation for the state support of young Russian scientists (Project No. MK-387.2012.3).References
- C. J. Suckling, in Trends in Medicinal Chemistry, ed. H. van der Goot, G. Domany, L. Pallos and H. Timmerman, Elsevier, Amsterdam, 1989, p. 805 Search PubMed.
- (a) E. Maquoi, N. E. Sounni, L. Devy, F. Olivier, F. Frankenne, H.-W. Krell, F. Grams, J.-M. Foidart and A. Noel, Clin. Cancer Res., 2004, 10, 4038 CrossRef CAS; (b) H.-J. Breyholz, S. Wagner, A. Faust, B. Riemann, C. Hoeltke, S. Hermann, O. Schober, M. Schafers and K. Kopka, Chem. Med. Chem., 2010, 5, 777 CrossRef CAS; (c) P. Bartsch, D. Cataldo, R. Endele, B. Evrard, J.-M. Foidart, H.-W. Krell and G. Zimmermann, Int. Appl. No. PCT EP, 2005, 003348 Search PubMed; (d) F. Grams, H. Brandstetter, S. D'Alo, D. Geppert, H.-W. Krell, H. Leinert, V. Livi, E. Menta, A. Oliva and G. Zimmermann, Biol. Chem., 2001, 382, 1277 CAS.
- (a) S. B. King, E. S. Statford, C. R. Craig and E. K. Fifer, Pharm. Res., 1995, 12(8), 1240 CrossRef CAS; (b) R. J. Prankerd and R. H. Mckeown, Int. J. Pharm., 1982, 83, 39 CrossRef; (c) L. Lomlin, J. Einsiedel, F. W. Heinemann, K. Meyer and P. Gmeiner, J. Org. Chem., 2008, 73, 3608 CrossRef.
- E. M. Galati, M. T. Monforte, N. Miceli and E. Ranerill, Farmaco, 2001, 56, 459 CrossRef CAS.
- S.-H. Kim, A. T. Pudzianowski, K. J. Leavitt, J. Barbosa, P. A. McDonnell, W. J. Metzler, B. M. Rankin, R. Liu, W. Vaccaro and W. Pitts, Bioorg. Med. Chem. Lett., 2005, 15, 1101 CrossRef CAS.
- W. Fraser, C. J. Suckling and H. C. S. Wood, J. Chem. Soc., Perkin Trans. 1, 1990, 3137 RSC.
- J. Duan, B. Jiang, L. Chen, Z. Lu, J. Barbosa and W. J. Pitts, US Pat. Appl., 2003, 0229084 Search PubMed.
- R. H. McKeown and R. J. Prankerds, J. Chem. Soc. Perkin Trans. 2: Phys. Org. Chem., 1981, 481 RSC.
- A. F. A. Shalaby and I. I. Abd El-Gawad, J. Prakt. Chem., 1971, 313, 1022 CrossRef CAS.
- S. Palwinder and P. Kamaldeep, J .Heterocyclic Chem., 2006, 607 Search PubMed.
- M. T. Maghsoodlou, S. M. H. Khorassani, R. Heydari, F. R. Charati, N. Hazeri, M. Lashkari, M. Rostamizadeh, G. Marandi, A. Sobolev and M. Makha, Tetrahedron Lett., 2009, 50, 4439 CrossRef CAS.
- (a) H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977 CrossRef CAS; (b) H. Pellissier, Tetrahedron, 2008, 64, 7041 CrossRef CAS; (c) A. Russo, S. Meninno, C. Tedesco and A. Lattanzi, Eur. J. Org. Chem., 2011, 5096 CrossRef CAS.
- (a) D. Kawai, K. Kawasumi, T. Miyahara, T. Hirashita and S. Araki, Synlett, 2008, 2977 CAS; (b) D. Kawai, K. Kawasumi, T. Miyahara, T. Hirashita and S. Araki, Tetrahedron, 2009, 65, 10390 CrossRef CAS.
- M. N. Elinson, A. N. Vereshchagin, N. O. Stepanov, T. A. Zaimovskaya, V. M. Merkulova and G. I. Nikishin, Tetrahedron Lett., 2010, 51, 428 CrossRef CAS.
- (a) H. Lund, Organic Electrochemistry, Marcel Dekker Inc., New York, 4th edn, 2000 Search PubMed; (b) S. Torii, Novel Trends in Electroorganic Synthesis, Springer, Berlin, 1998 Search PubMed.
- (a) T. Shono, Electroorganic Chemistry As a New Tool in Organic Synthesis, Springer, Berlin, 1994 Search PubMed; (b) Y. N. Ogibin, M. N. Elinson and G. I. Nikishin, Russ. Chem. Rev., 2009, 78, 89 CrossRef CAS; (c) G. I. Nikishin, M. N. Elinson and I. V. Makhova, Angew. Chem., 1988, 100, 1782 CrossRef; (d) M. N. Elinson, A. S. Dorofeev, S. K. Feducovich, R. F. Nasybullin, E. F. Litvin, M. V. Kopyshev and G. I. Nikishin, Tetrahedron, 2006, 62, 8021 CrossRef CAS; (e) M. N. Elinson, A. S. Dorofeev, S. K. Feducovich, P. A. Belyakov and G. I. Nikishin, Eur. J. Org. Chem., 2007, 3023 CrossRef CAS; (f) M. N. Elinson, A. I. Ilovaisky, V. M. Merkulova, T. A. Zaimovskaya, P. A. Belyakov and G. I. Nikishin, Mendeleev Commun., 2010, 20, 207 CrossRef CAS.
- For selected publications, see: (a) M. N. Elinson, S. K. Feducovich, Z. A. Starikova, O. S. Olessova, A. N. Vereshchagin and G. I. Nikishin, Tetrahedron Lett., 2000, 41, 4937 CrossRef CAS; (b) M. N. Elinson, S. K. Feducovich, Z. A. Starikova, A. N. Vereshchagin, P. A. Belyakov and G. I. Nikishin, Tetrahedron, 2006, 62, 3989 CrossRef CAS; (c) M. N. Elinson, S. K. Feducovich, A. N. Vereshchagin, S. V. Gorbunov, P. A. Belyakov and G. I. Nikishin, Tetrahedron Lett., 2006, 47, 9129 CrossRef CAS; (d) A. N. Vereshchagin, M. N. Elinson, T. A. Zaimovskaya and G. I. Nikishin, Tetrahedron, 2008, 64, 9766 CrossRef CAS.
- F. Seeliger, S. T. A. Berger, G. Y. Remennikov, K. Polborn and H. Mayr, J. Org. Chem., 2007, 72, 9170 CrossRef CAS.
- The described observation has prompted us to conduct the following study on electrocatalytic synthesis of pyrano[2,3-d]pyrimidines: M. N. Elinson, A. I. Ilovaisky, V. M. Merkulova, T. A. Zaimovskaya and G. I. Nikishin, Mendeleev Commun., 2011, 21, 122 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112 CrossRef.
Footnote |
| † CCDC reference number 855338. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra20078c |
| This journal is © The Royal Society of Chemistry 2012 |