NOx storage and reduction with methane by plasma at ambient temperature†
Hui
Wang
,
Qinqin
Yu
,
Tong
Liu
,
Liping
Xiao
and
Xiaoming
Zheng
*
Key Lab of Applied Chemistry of Zhejiang Province, Zhejiang University, Hangzhou 310028, China. E-mail: xmzheng@zju.edu.cn; Fax: (+)86 571 88273283; Tel: (+)86 571 88273417
First published on 4th April 2012
Abstract
NOx storage and reduction with CH4 by a plasma process was proposed for NOx removal at ambient temperature. The efficiency of this new process for NOx removal could achieve 95% at ambient temperature. NOx removal via cyclic operation has also been investigated, maintaining efficieniency above 90%.
In the light of stringent world-wide environmental legislation, nitrogen oxide NOx (NO, NO2, N2O), one of the most dangerous gaseous pollutants, requires new abatement technologies.1 Selective catalytic reduction using methane as a reducing agent (CH4-SCR) has been studied as a desirable technology for removal of NOx in the presence of excess oxygen for a long time.2 CH4 has attracted considerable attention because of its vast reserves, low cost and easy handling. However, it is difficult to activate CH4 for the high stability of the C–H bonds. To reduce the reaction activation energy through catalysis is one of the main challenges in CH4–SCR. Unfortunately, the existing catalysts, even modified by noble metal such as Pd/Co/mordenite,3 Pt/Co/mordenite4 and Pt/Co/ZSM-5,5 exhibited satisfactory activity for CH4–SCR only when the temperature was more than 673 K. High reaction temperature means high energy consumption, which is unacceptable considering industrial applications, especially for stationary sources. It is hence meaningful to find a solution that can improve the low-temperature activity of the CH4–SCR.
Non-thermal plasma (NTP) could activate molecules including NOx and hydrocarbons at ambient temperature. Thus, there has been extensive research on NTP for NOx removal recently. Plasma procedures for NOx removal mainly include plasma-assisted SCR6 and plasma-derived NO decomposition process.7 Nonetheless, it requires high energy-consumption to remove NOx at low concentration from exhaust gas or flue gas by constant plasma discharge. To solve the problem, an improved plasma process was proposed by several groups.8 The pollutant is first enriched on the adsorbents, and then the adsorbed pollutant is desorbed or decomposed by plasma in a short time. But an effective de-NOx by plasma is barely possible in the absence of an oxygen atmosphere.9 O2 derived from carrier gas (air or combustion gas) and NOx decomposed can react with N2 to generate a considerable amount of NOx in plasma. Briefly, the reverse reaction of NOx decomposition leads to a reduction of the total de-NOx efficiency.
Deriving from the traditional NOx storage and reduction (NSR) technology10 combined NTP, we reported herein a NSR process in plasma using CH4 as a reducing agent. First, diluted NOx was on using commercial H-ZSM-5 (Si/Al ratio at 22, without further modification) for a long time. Then, the adsorbed pollutant was reduced with CH4 by NTP in a few minutes. Notably, in the discharge stage, we chose combustion gas (oxygen-deficient gas containing nearly 5% O2) instead of air as a carrier gas. Moreover, the whole cyclic process carried out at ambient temperature. Finally, a high-efficient process for NOx removal was obtained at low energy consumption, even without the use of any noble metal.
Fig. 1 shows the schematic diagram of the combined CH4-NSR and plasma system. The dielectric barrier discharge (DBD) reactor consists of a quartz tube (i.d. 8.5 mm) as the reactor as well as the dielectric barrier, a stainless steel rod (o.d. 3.5 mm) as the high-voltage electrode, and a stainless steel wire mesh wrapped on the outside of the quartz tube as the grounding electrode. H-ZSM-5 was filled in the discharge gap between the high-voltage electrode and the quartz tube. The DBD reactor was energized by an AC power source (Nanjing Suman Electronics Co. Ltd). The waveforms of the discharge current and the applied voltage were monitored with a digital oscilloscope (Tektronix, TDS 1002B-SC). Discharge power was measured by the V–Q Lissajous program.
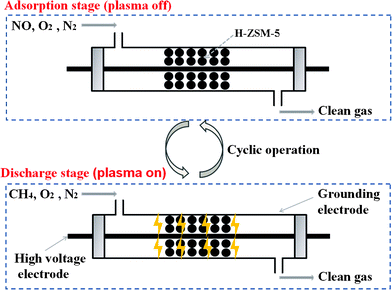 | ||
| Fig. 1 Schematic diagram of the combined CH4–NSR and plasma system. | ||
Fig. 2 (a) shows the conversion of the adsorbed NOx to N2 on H-ZSM-5 in CH4–NSR by plasma as a function of discharge power. H-ZSM-5 acted as an adsorbent for NOx at the first stage and a catalyst at the second stage owing to its acid site. Clearly, the conversion of adsorbed NOx to N2 improved greatly with increasing discharge power, reaching 95% at 6.7 W. High energy density is beneficial to NOx conversion in CH4–NSR, according with the results reported in other reaction systems.11 Moreover, CH4 conversion remains 100% at different discharge power during the reaction as shown in Fig. 2 (b). And the selectivity of CH4 to CO2 was also nearly 100%, because CO was easily oxidized to CO2 in plasma. At the same time, no NOx was detected on the adsorbent at each discharge power after the discharge stage through a temperature-programmed desorption (TPD) process. Fig. 2 (c) shows that NOx conversions in NOx decomposition without CH4 are all below 40% due to the reverse reaction of NOx decomposition (NOx formation reaction). Obviously, CH4 enhanced the total NOx conversion. On one hand, a suitable amount of CH4 could play a role as a reducing agent in NSR; on the other hand, CH4 oxidation consumed O2 to restrain the reverse reaction of NOx decomposition effectively. On-line mass spectrometry (MS) detected no N2O5, N2O4, or N2O3 produced during the reaction. Quantitative analysis indicated that the selectivity of NOx to N2O was lower than 0.5% in CH4-NSR.
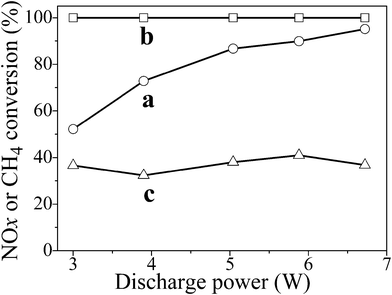 | ||
| Fig. 2 (a) Conversion of adsorbed NOx to N2 and (b) CH4 to CO2 in CH4-NSR and (c) conversion of adsorbed NOx in decomposition of NOx without CH4 as a function of discharge power. Conditions: 1 g H-ZSM-5, 30 mL min−1 flow rate of oxygen-deficient air (5% O2 + 95% N2) with or without 1.67% CH4, total input energy of the plasma (P×tdischarge) is fixed at 0.896 W h. | ||
The direct decomposition of NO (gaseous phase) in plasma has been investigated. Herein, through NO decomposition, we deduced fundamental rules and phenomenon about NO abatement (decomposition or reduction) in plasma. The result is shown in Fig. 3 (A). As the discharge power increased, the decomposition of 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm NO at high flow rate (80 mL min−1) moved up significantly, achieving more than 99% at 6.72 W. This phenomenon suggested that high energy density led to high NOx conversion once again. When the total flow rate decreased to 30 mL min−1 to enhance the energy density of system, NO decomposed to N2 almost 100% at all discharge power except for the lowest, 3 W. Reducing NO concentration (2000 ppm or 10000 ppm) in the feed to make further improvement of energy density, the decomposition of NO was 100% even at 3 W. Fig. 3 (B) displays the effect of O2 concentration and discharge power on the reverse reaction of NOx decomposition (NOx formation reaction). The yield of NOx increased rapidly with increasing the O2 concentration. Interestingly, the discharge power did not affect the yield of NOx, suggesting that energy density did not affect the reverse reaction of NOx decomposition. Based on the above analysis, the phenomenon shown in Fig. 2 could be explained. In order to improve the NOx conversion, O2 concentration in the system must be controlled by introducing of CH4 in the feed. Enhancing discharge power to raise the energy density of system was beneficial for NOx removal and did not affect the corresponding reverse reaction.
000 ppm NO at high flow rate (80 mL min−1) moved up significantly, achieving more than 99% at 6.72 W. This phenomenon suggested that high energy density led to high NOx conversion once again. When the total flow rate decreased to 30 mL min−1 to enhance the energy density of system, NO decomposed to N2 almost 100% at all discharge power except for the lowest, 3 W. Reducing NO concentration (2000 ppm or 10000 ppm) in the feed to make further improvement of energy density, the decomposition of NO was 100% even at 3 W. Fig. 3 (B) displays the effect of O2 concentration and discharge power on the reverse reaction of NOx decomposition (NOx formation reaction). The yield of NOx increased rapidly with increasing the O2 concentration. Interestingly, the discharge power did not affect the yield of NOx, suggesting that energy density did not affect the reverse reaction of NOx decomposition. Based on the above analysis, the phenomenon shown in Fig. 2 could be explained. In order to improve the NOx conversion, O2 concentration in the system must be controlled by introducing of CH4 in the feed. Enhancing discharge power to raise the energy density of system was beneficial for NOx removal and did not affect the corresponding reverse reaction.
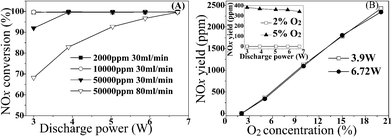 | ||
| Fig. 3 (A) NO decomposition in plasma as a function of discharge power. (B) NOx yield in the reaction of O2 with N2 as a function of O2 concentration and discharge power (inset) in plasma. Conditions A: various concentration NO (balanced N2) with a certain flow rate in the absence of adsorbent. Conditions B: 30 mL min−1 of total flow rate, discharge power is 6.72 W in the absence of adsorbent. | ||
In order to further optimize the discharge process, we investigated the effect of flow rate and CH4/O2 molar ratio on the conversion of adsorbed NOx to N2 in the discharge stage. Fig. 4 (a) shows that NOx conversion decreased with increased flow rate. When the flow rate increased, the energy density of the system decreased at the same discharge power. Low energy density led to low NOx conversion, in agreement with the result from Fig. 2 (a). Actually, the demand for lower flow rate at the discharge stage will never be a drawback to this proposed technology, because the discharge time is very short. In addition, low flow rate can cut down the consumption of the reducing agent CH4 for NSR. On the other hand, the adsorption stage, for reaction long times, is not affected by high flow rate due to the superior storage performance of H-ZSM-5 for NOx. Fig. 4 (b) displays NOx conversion as a function of CH4/O2 molar ratio. The positive effect of CH4 added in the feed was remarkable. The conversion of NOx to N2 was significantly higher (more than 95%) when the CH4/O2 molar ratio was between 0.333 and 1. When the CH4/O2 molar ratio exceeded 1, the NOx conversion fell off and an excess of CH4 also could bring into the greenhouse effect.
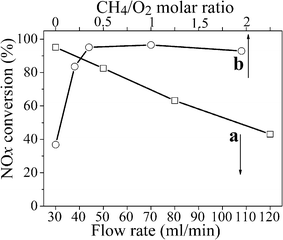 | ||
| Fig. 4 Effect of (a) flow rate and (b) CH4/O2 molar ratio on the conversion of adsorbed NOx in CH4-NSR. Conditions a: 1 g H-ZSM-5, different flow rate of oxygen-deficient air (5% O2 + 95% N2) with 1.67% CH4, total input energy of the plasma is fixed at 6.72 W × 8 min. Conditions b: 1 g H-ZSM-5, 30 mL min−1 flow rate of oxygen-deficient air (5% O2 + 95% N2) with various CH4/O2 molar ratios, total input energy of the plasma is fixed at 6.72 W × 8 min. | ||
Cyclic operation of the present process for NOx removal has been investigated (Fig. 5). Notably, between the intermissions of two cycles, the absorbent H-ZSM-5 does not need to be treated for regeneration in any way. Over the adsorption stage, a large amount of NOx could be completely trapped on H-ZSM-5, due to no NOx remaining on the adsorbent after the discharge stage. In the discharge stage, the conversion of adsorbed NOx to N2 decreased a little in the second time, and then kept at above 90% in continuing reaction cycles. NOx removal of cyclic operation was satisfactory. XRD patterns of H-ZSM-5 before and after reaction (Fig. 1S in the ESI†) showed that H-ZSM-5 still kept its crystalline structure during the cyclic reaction. Both results suggested that the performance of H-ZSM-5 is stable under the process conditions.
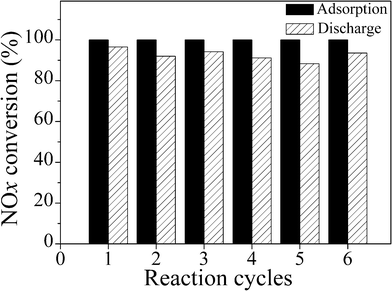 | ||
| Fig. 5 Cyclic operation of the present adsorption–discharge process for NOx removal: percentage of NOx abatement in the adsorption stage (black) and conversion of adsorbed NOx to N2 in the discharge stage (slash).Conditions: in the adsorption stage, 1 g H-ZSM-5 adsorbed 66 mL min−1 flow rate containing 1840 ppm NO (8% O2, balanced N2) for 1 h; in the discharge stage, 30 mL min−1 flow rate of oxygen-deficient air (5% O2 + 95% N2) with 5% CH4, total input energy of the plasma is fixed at 6.72 W × 7 min = 0.784 W h. | ||
In conclusion, the CH4-NSR process by plasma could remove NOx efficiently achieving 95%removal at ambient temperature. Plasma primarily activated the gaseous molecule. CH4 introduced into the system played an important role as a reducing agent and also restrained the reverse reaction of NOx decomposition effectively. A high energy efficiency for the removal of NOx was obtained, 0.416 mmol NOx/W h (ESI†), due to the long-time adsorption and short-time discharge in the process. The energy efficiency of the new process proposed was improved, compared with the conventional thermal catalytic process. Thus, we believe that this new de-NOx technology has broad application prospects.
Experimental section
Before the experiment, 1 g H-ZSM-5 (Si/Al = 22) (20–40 mesh) was pretreated at 773 K for 60 min in N2 flow, and then cooled down to room temperature. The whole experiment consisted of two stages. In the adsorption stage (plasma off), a flowing mixed gas containing 1840 ppm NO and 8% O2 balanced N2 was introduced into the catalyst-packed dielectric barrier discharge (DBD) reactor at a total flow rate of 66 mL min−1. This stage lasted for 1 h at 308 K and H-ZSM-5 absorbed 0.326 mmol NOx. In the discharge stage (plasma on), the gas stream oxygen-deficient air (5% O2 + 95%N2) and a small quantity of CH4 was switched into the DBD reactor. This discharge process just lasted several minutes at room temperature. The total input energy of the plasma (P×tdischarge) is fixed at 0.896 W h (except the cyclic experiment).The concentrations of NO and NO2 were analyzed online by a chemiluminescent NO/NO2/NOx analyzer (Eco Physics). N2O was analyzed using a Fuli-9750 gas chromatograph (GC) with TCD and a Porapak Q column. The definitions of the conversion of NOx to N2 and energy efficiency were defined as follows:
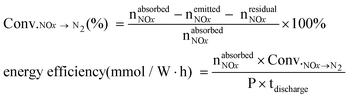 | (1) |
Acknowledgements
This work is financially supported by Major project of Zhejiang Province (No.2009C11141), the National Natural Science Foundation of China (No.20977080) and the Natural Science Foundation of Zhejiang Province (No.J20091504).References
- (a) P. Forzatti, I. Nova and E. Tronconi, Angew. Chem., 2009, 121, 8516 CrossRef; (b) C. Petitto, H. P. Mutin and G. Delahay, Chem. Commun., 2011, 47, 10728 RSC.
- (a) M. C. Campa, S. De Rossi, G. Ferraris and V. Indovina, Appl. Catal., B, 1996, 8, 315 CrossRef CAS; (b) L. Gutierrez and E. A. Lombardo, Appl. Catal. A, 2009, 306, 107 CrossRef.
- (a) F. Bustamante, F. Córdoba, M. Yates and C. M. de Correa, Appl. Catal., A, 2002, 234, 127 CrossRef CAS; (b) L. F. Córdoba, G. A. Fuentes and C. M. de Correa, Microporous Mesoporous Mater., 2005, 77, 193 CrossRef.
- (a) L. Gutierrez, A. V. Boix, E. A. Lombardo and J. L. G. Fierro, J. Catal., 2001, 199, 60 CrossRef CAS; (b) L. Gutierrez, M. A. Ulla, E. A. Lombardo, A. Kovacs, F. Lonyi and J. Valyon, Appl. Catal., A, 2005, 292, 154 CrossRef CAS.
- L. Gutierrez, A. Boix and J. O. Petunchi, J. Catal., 1998, 179, 179 CrossRef CAS.
- (a) D. N. Tran, C. L. Aardahl, K. G. Rappe, P. W. Park and C. L. Boyer, Appl. Catal., B, 2004, 48, 155 CrossRef CAS; (b) J. L. Hueso, J. Cotrino, A. Caballero, J. P. Espinós and A. R. González-Elipe, J. Catal., 2007, 247, 288 CrossRef CAS.
- (a) J. Luo, S. L. Suib, M. Marquez, Y. Hayashi and H. Matsumoto, J. Phys. Chem. A, 1998, 102, 7954 CrossRef CAS; (b) S. L. Suib, S. L. Brock, M. Marquez, J. Luo, H. Matsumoto and Y. Hayashi, J. Phys. Chem. B, 1998, 102, 9661 CrossRef CAS.
- (a) M. Okubo, G. Tanioka, T. Kuroki and T. Yamamoto, IEEE Trans. Ind. Appl., 2002, 38, 1196 CrossRef CAS; (b) H. H. Kim, A. Ogata and S. Futamura, Appl. Catal., B, 2008, 79, 356 CrossRef CAS; (c) Q. Q. Yu, H. Wang, T. Liu, L. P. Xiao, X. Y. Jiang and X. M. Zheng, Environ. Sci. Technol., 2012, 46, 2337 CrossRef CAS.
- M. Baeva, H. Gier, A. Pott, J. Uhlenbusch, J. Hoschele and J. Steinwandel, Plasma Sources Sci. Technol., 2002, 11, 1 CrossRef CAS.
- (a) L. Castoldi, I. Nova, L. Lietti and P. Forzatti, Catal. Today, 2004, 96, 43 CrossRef CAS; (b) S. Roy and A. Baiker, Chem. Rev., 2009, 109, 4054 CrossRef CAS.
- (a) A. Ogata, H. Einaga, H. Kabashima, S. Futamura, S. Kushiyama and H. H. Kim, Appl. Catal., B, 2003, 46, 87 CrossRef CAS; (b) A. M. Harling, D. J. Glover, J. C. Whitehead and K. Zhang, Ind. Eng. Chem. Res., 2008, 47, 5856 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20115a/ |
| This journal is © The Royal Society of Chemistry 2012 |