Tuning the bactericidal repertoire and potency of quinoline-based amphiphiles for enhanced killing of pathogenic bacteria†
Umakanth
Vudumula
a,
Manab Deb
Adhikari
a,
Bimlesh
Ojha
b,
Sudeep
Goswami
a,
Gopal
Das
*b and
Aiyagari
Ramesh
*a
aDepartment of Biotechnology, Indian Institute of Technology Guwahati, Guwahati, 781039, India. E-mail: aramesh@iitg.ernet.in; Fax: +91 361 2582249; Tel: +91 361 2582205
bDepartment of Chemistry, Indian Institute of Technology Guwahati, Guwahati, 781039, India. E-mail: gdas@iitg.ernet.in; Fax: + 91 361 2582349; Tel: +91 3612582313
First published on 9th March 2012
Abstract
The overwhelming challenge posed by drug-resistant pathogenic bacteria underscores the need for potent bactericidal agents, which exhibit broad-spectrum activity and a mode of action that does not favor development of resistance. In the present study we report the synthesis and bactericidal activity of structurally diverse quinoline-based amphiphiles, having a fluorescent head group and varying hydrophobic chain length. A structure-guided bactericidal efficacy and broad-spectrum activity of the amphiphiles was apparent in screening experiments against a panel of common pathogenic bacteria. Structure–function studies by fluorescence-based assays revealed that the charge and hydrophobic chain length of amphiphiles were key structural determinants that radically boosted the bactericidal activity. The most potent amphiphile N-methyl 8-dodecoxy quinolinium iodide (compound 6) exhibited a dose-dependent bactericidal activity on target pathogens and could even inhibit the growth of a presumptive methicillin-resistant S. aureus (MRSA) strain. Fluorescence-based mechanistic studies and transmission electron microscope (TEM) analysis indicated that the initial binding of compound 6 to bacteria probably involved electrostatic interaction, whereas the hydrophobic chain of the amphiphile promoted membrane insertion, which culminated in large scale membrane disruption and loss in cell viability. Although the bactericidal activity of compound 6 was independent of bacterial transmembrane potential, interaction of the amphiphile with pathogenic bacteria resulted in rapid dissipation of membrane potential. Interestingly, compound 6 displayed high antimicrobial selectivity and did not affect the viability of human HT-29 cells. It is envisaged that the therapeutic regime of the bactericidal scaffold of compound 6 can be further expanded by rational structural design for generating potent bactericidal agents.
1 Introduction
The emergence of drug-resistant pathogenic bacteria as a result of indiscriminate and arbitrary use of antibiotics is a serious healthcare concern. This is compounded by the fact that a gamut of elegant molecular mechanisms has empowered pathogenic bacteria to develop resistance against conventional drugs.1–4 Clearly, new antibiotics are the need of the hour to combat these emerging pathogens, but progress in discovery and development of novel antibiotic-based therapeutic regimes has been rather slow.5 In the quest for novel bactericidal agents, the current interest is in molecules that are potent, broad-spectrum and with a mechanism of action that does not readily favor development of resistance. In this regard, antimicrobial agents that target critical entities in pathogenic bacteria, such as membrane function, hold considerable promise since the probability of developing resistance against such compounds would necessitate restoration of membrane components, a task which is physiologically daunting for the bacteria.6,7Amongst the present antimicrobial agents, antimicrobial peptides (AMPs) are attractive. Structure–function studies have clearly demonstrated that positively charged residues of AMPs initiate interactions with the negatively charged cell surface of bacteria and hydrophobicity facilitates their insertion into the hydrophobic core of membrane resulting in cell disruption.8–11 In spite of the promise as bactericidal agents, the therapeutic potential of AMPs is hampered owing to high manufacturing cost, poor pharmacokinetics and the possibility of resistance development in target pathogens.11,12
The current thrust in medicinal chemistry research has been in the synthesis and characterization of drug scaffolds, which mimic AMPs in their mechanism of action. In the context of bio-mimetic candidate molecules, synthetic amphiphilic compounds have attracted enormous scientific interest, owing to their facile synthesis, their capacity to act on bacterial cell membrane and the possibility of modulating their activity by subtle alterations in structure. A large number of studies demonstrate potent antibacterial activity of various classes of amphiphilic compounds against pathogenic and drug-resistant bacterial strains.13–18 A common theme in the mechanism of action of synthetic amphiphiles is that they are membrane acting and their antibacterial activity is largely influenced by cationic charge as well as hydrophobicity of the molecule.9,15,19–21 With regard to the therapeutic application of synthetic amphiphiles as antimicrobial agents, assessment of cytotoxicity is critical and a number of studies have stressed the importance of antimicrobial selectivity and reduced cytotoxicity towards human cells.14,15,20,22
Our research group has been actively involved in the synthesis of amphiphiles, characterization of their photophysical properties and interaction with biomolecules for protein sensing and inhibition of metalloenzymes.23–27 In continuation of our interest in biological application of synthetic amphiphiles and development of broad-spectrum antimicrobial agents in particular, we report the synthesis of neutral and cationic quinoline-based amphiphilic compounds with varying alkyl chain length and their antibacterial activity against a set of common pathogenic bacteria. A systematic structure–function study for the rational selection of the most potent amphiphile and the mode of action and cytotoxicity study of this amphiphile are also reported in the present work.
2 Experimental
Neutral amphiphiles were prepared following the literature method.28 Detailed synthesis and characterization of cationic amphiphiles is described in the ESI†. The general structure of the amphiphiles is shown in Fig. 1.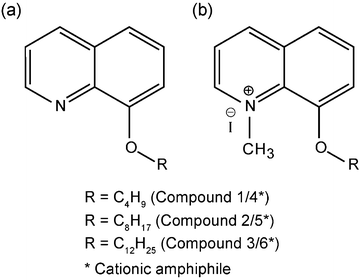 | ||
| Fig. 1 General structure of synthetic amphiphiles used in the study. (a) 8-Alkoxy quinoline (neutral) and (b)N-methyl 8-alkoxy quinolinium iodide (cationic). | ||
Antibacterial activity of the amphiphiles was tested against Gram-positive Bacillus subtilis MTCC 441 (B. subtilis), Listeria monocytogenes Scott A (L. monocytogenes), Staphylococcus aureus MTCC 96 (S. aureus) and Gram-negative Escherichia coli MTCC 433 (E. coli), Enterobacter aerogenes MTCC 2822 (E. aerogenes) and Pseudomonas aeruginosa MTCC 2488 (P. aeruginosa). Minimum inhibitory concentration (MIC) and minimum killing concentration (MKC) of the synthetic amphiphiles were determined against E. coli MTCC 433 and S. aureus MTCC 96 by a broth dilution method. Structure–function studies on amphiphiles were pursued using a fluorescence-based assay. The dose-dependent bactericidal activity of compound 6 was determined by estimating viable cells using a conventional serial dilution and plating method.
The mode of action of compound 6 on pathogenic bacteria was determined using fluorescence-based assays as well as transmission electron microscope (TEM) analysis. Cytotoxicity of compound 6 was assessed on human HT-29 colon adenocarcinoma cells by a standard XTT assay following the manufacturer’s instructions (Sigma-Aldrich, MO, USA).
A detailed description of all the above-mentioned experimental procedures is available in the ESI†.
3 Results and discussion
The ever increasing menace of drug-resistant pathogenic bacteria has accentuated the need for novel and potent bactericidal agents. In this context, antimicrobial peptides (AMPs) constitute a diverse class of antibacterial agents that hold considerable promise. Characteristic structural attributes of these molecules, in particular their amphiphilic topology contributes to the high propensity of membrane insertion, which results in rapid killing of target bacteria. This mode of action is highly desirable in an antimicrobial agent as probability of development of resistance is quite unlikely. However, large scale exploitation of AMPs is precluded by the high manufacturing costs, poor pharmacokinetics and low bactericidal efficacy. On the other hand, bio-inspired synthetic amphiphiles that mimic the bactericidal scaffold of AMPs could provide a plethora of diverse lead compounds owing to their facile synthesis, superior pharmacokinetic attributes and high bactericidal activity. This provided the motivation for our work wherein we synthesized structurally diverse quinoline-based amphiphiles and investigated the structure-guided antimicrobial efficacy and mode of action of the amphiphile on pathogenic bacteria.3.1 Antibacterial activity of synthetic amphiphiles
The design strategy for the amphiphiles was based on: (a) incorporation of a head group that can be readily converted from neutral to cationic counterpart, (b) incorporation of a fluorophore unit in the head group to generate an efficient sensing element and (c) varying the hydrophobic chain length for modulating membrane insertion property. The results of the bactericidal activity with 100 μg mL−1 neutral amphiphiles are indicated in Fig. 2a. When compound 1, having the shortest chain length, was tested, marginal growth inhibition (around 84% growth compared to control) was observed in the case of P. aeruginosa, E. aerogenes and L. monocytogenes. With a higher alkyl chain length of the amphiphile (compound 2), the growth of these pathogens was further suppressed (around 70–73% compared to control). Growth inhibition for the same pathogens was more prominent with compound 3 (having the highest alkyl chain length) as compared to compound 1 and compound 2. It is also noticeable that upon exposure to compound 3, growth inhibition was also observed to some extent for the pathogens S. aureus, E. coli and B. subtilis. Amongst the neutral amphiphiles, the approximate hydrophobicity (LogP value) of compound 3 was highest followed by compound 2 and compound 1, respectively (refer to ESI†, Table S1), which indicates that the bactericidal potency of the neutral amphiphiles is probably correlated to an increase in hydrophobicity.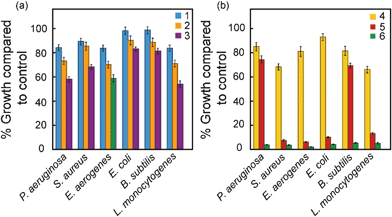 | ||
| Fig. 2 Antimicrobial activity of (a) neutral (compounds 1, 2 and 3) and (b) charged amphiphiles (compounds 4, 5 and 6) against pathogenic bacterial strains. | ||
Bacterial cells are known to be negatively charged owing to the presence of teichoic acid in Gram-positive bacteria and lipopolysaccharides in Gram-negative bacteria.29,30 We conceived that imparting a positive charge to the same set of amphiphiles would result in enhanced electro-affinity for the bacterial cell surface and lead to broad-spectrum bactericidal activity since the interaction is based on a ubiquitous charge characteristic of bacterial cells. It can be seen from Fig. 2b that in case of compound 4, the bactericidal effect on the pathogens was superior to that observed for the corresponding neutral amphiphile (compound 1). This difference was especially noticeable in the case of Gram-positive pathogens S. aureus and L. monocytogenes, wherein the growth compared to control was around 68% and 66%, respectively. Treatment of bacterial cells with compound 5, having a higher alkyl chain length, resulted in an increased spectrum of activity and virtually complete growth inhibition for S. aureus, E. aerogenes, E. coli and L. monocytogenes (Fig. 2b). When the bactericidal effect of the cationic amphiphile having the highest alkyl chain length (compound 6) was tested, the outcome was remarkable as growth of all the pathogens was completely abolished (Fig. 2b). Hence introduction of a positive charge in the amphiphile significantly improved its bactericidal efficacy and an increase in alkyl chain length of the cationic amphiphile dramatically enhanced the bactericidal efficacy as well as the spectrum of activity against pathogenic bacteria. A similar trend was also observed at 50 μg mL−1 amphiphile concentration (refer to ESI†, Fig. S1). The cationic nature of the amphiphile may promote the initial interaction with the negatively charged bacterial cell surface, as suggested earlier for antimicrobial peptides.31 On the other hand, the hydrophobic tail of the amphiphile may enhance its affinity for the hydrophobic core of a bacterial cell membrane, steer its insertion into the membrane and eventually cause membrane disruption. This fundamental tenet has been reiterated in earlier reports on amphiphilic antimicrobial agents.15,21,32 The key role of hydrophobicity of the amphiphile in the context of antimicrobial activity is explicit in our results and a striking increase in antibacterial activity with increase in hydrophobic tail length is consistent with earlier reports on polymeric and peptide-based antimicrobial agents.9,15,19,33 Control experiments revealed that individual functional entities of the amphiphile (iodide, hydroxyl quinolinium or the alkyl chain) failed to inhibit the growth of E. coli and S. aureus when tested separately (ESI†, Fig. S2). It may be mentioned that the concentrations of these components were equivalent to those present in 100 μg mL−1 (220 μM) of compound 6.
Interestingly, a disc diffusion assay revealed that compound 6 could arrest the growth of the Gram-positive pathogen S. aureus MTCC 96 and S. aureus MTCC 740 (ESI†, Fig. S3), which were identified as presumptive methicillin-resistant S. aureus (MRSA) and methicillin-sensitive S. aureus (MSSA), respectively, following the recommended protocol of the Clinical and Laboratory Standards Institute.34S. aureus infection in community and hospital settings has serious healthcare implications. The pathogen is known to cause a wide range of disease, ranging from food poisoning, device and wound-related infections to life-threatening ailments. MRSA strains are of particular concern as they are resistant to β-lactams and other antibiotics commonly used to control S. aureus infection.35 To curb the menace of this pathogen, alternate targets as well as novel antibacterial agents are being explored.6,36 In this context, the potential of compound 6 to suppress the growth of strain MTCC 96, a presumptive MRSA, appears promising. It may be mentioned that the strain MTCC 96 is procured from a culture collection centre, and is a reference strain used for antimicrobial susceptibility tests. It would be interesting to test the efficacy of compound 6 against clinical isolates of MRSA.
3.2 Minimum inhibitory concentration (MIC) and minimum killing concentration (MKC)
The MIC and MKC of the amphiphiles were determined for E. coli and S. aureus (refer to ESI†, Table S2). Compound 6 demonstrated the most potent antibacterial activity against the target pathogens. The MIC and MKC for this amphiphile was 15 and 20 μM, respectively, for E. coli, whereas for S. aureus it amounted to 5 and 15 μM, respectively.3.3 Structure–activity relationship
Fluorescence-based experiments were conducted to probe the structure–function relationship for compounds 1, 3 and 6, which differ in hydrophobicity and charge characteristics. In the first experiment, 5 (and 6)-carboxyfluorescein diacetate succinimidyl ester (cFDA-SE) labeled cells of S. aureus were treated with equimolar concentrations of the amphiphiles for 3 h and the extent of dye leakage from cells was compared to determine the antimicrobial efficacy of the amphiphiles. It is quite evident from Fig. 3a that treatment of S. aureus with 4.5 μM compound 6 resulted in the highest efflux of the dye (around 6.3 × 107 cps). At the same amphiphile concentration, the extent of dye leakage from the cells decreased progressively for compound 3 (4.5 × 107 cps) and compound 1 (1.7 × 107 cps). When a higher amphiphile concentration was used (7.5 μM) dye leakage from S. aureus was slightly higher in magnitude indicating a dose-dependent effect. Following uptake of cFDA-SE by cells of S. aureus and subsequent cleavage of the ester bond of the dye by intracellular esterase activity, the fluorescent form of the dye is likely irreversibly conjugated to intracellular and cell-surface proteins and accumulates in the cell.37 Hence it may be construed that in amphiphile-treated cells, leakage of the fluorescent dye is a consequence of extensive membrane damage. A distinct gradient in the extent of dye leakage from target cells treated with equimolar amphiphile concentrations, suggests that compound 6 exhibited the highest efficacy of membrane insertion and pore formation among the amphiphile series. Membrane damage in amphiphile-treated target cells was also determined by uptake of propidium iodide (PI), a cell-impermeant dye that enters cells only if the membrane is permeabilized or compromised.38 Upon entry into cells, PI binds to single- and double-stranded nucleic acids, yielding an intense red fluorescence. It is evident from Fig. 3b that the highest PI uptake was observed for cells treated with compound 6, indicating that this amphiphile caused maximum membrane damage. These results are consistent with the earlier results obtained for cFDA-SE leakage (Fig. 3a). Fluorescence microscopy provided additional evidence for structure-guided antimicrobial activity of the amphiphiles (Fig. 3c). A decrease in the number of cFDA-SE stained cells of S. aureus treated with compounds 1, 3 and 6, respectively, indicated a progressive loss in cell viability, as cFDA-SE fluorescence is based on intracellular esterase activity of viable cells. These results were also supported by large number of PI stained cells conspicuous in case of treatment with compound 6, indicating maximum membrane damage compared to untreated control cells as well as cells treated with other amphiphiles (compound 1 and 3). A similar trend was also observed for amphiphile-treated cells of E. coli when cFDA-SE leakage assay, PI uptake assay and fluorescence microscopic analysis was conducted (data not shown). Collectively, the results of the fluorescence-based experiments establish the structure–function relationship of the amphiphiles and are consistent with the trends reported earlier for other antimicrobial agents.8,9,15 Subsequent experiments were conducted with compound 6, which was evidently most potent amongst the amphiphiles.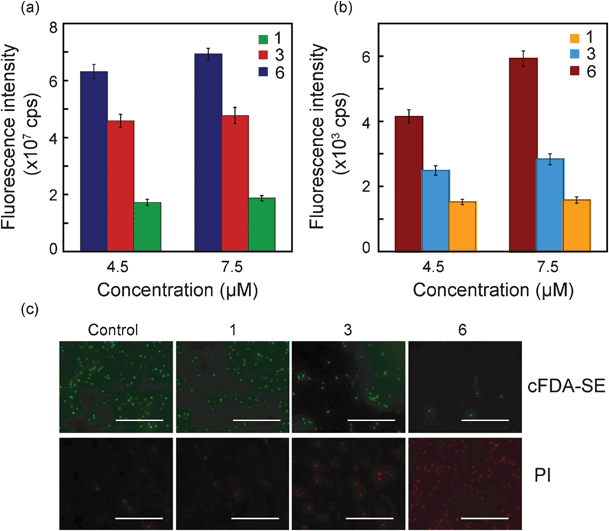 | ||
| Fig. 3 Structure–function studies for amphiphiles (compound 1, 3 and 6) on target bacteria S. aureus MTCC 96. (a) cFDA-SE leakage assay, (b) PI uptake assay and (c) Fluorescence microscopic images of amphiphile-treated cells labeled with cFDA-SE and PI. Scale bar for all the images is 50 μm. | ||
3.4 Bactericidal activity of compound 6
In these experiments, 106 CFU mL−1 of E. coli and S. aureus were suspended in PBS and treated with varying concentrations of compound 6. It may be mentioned that evaluation of the bactericidal activity of the amphiphile in a nutrient-free medium such as PBS is critical. Bacterial cells are less likely to proliferate in PBS owing to lack of essential nutrients. Hence, the results obtained in bactericidal assays performed in PBS would be independent of the ability of the bacterial strains to proliferate, and thus provide an authentic measure of the bactericidal efficacy of the amphiphile. As is evident from Fig. S4 (ESI†), there was a systematic decrease in cell viability with increase in amphiphile concentration and prolonged interaction time with the amphiphile. Bacterial cell viability was not affected in PBS and DMSO, indicating that the bactericidal effect was solely due to exposure of cells to amphiphile. Compound 6 was inherently fluorescent and thus its sequestration by bacterial cells could be pursued by allowing amphiphile–bacteria interactions in PBS followed by separation of cells by centrifugation and measuring fluorescence of the unbound amphiphile recovered in the supernatant. It was observed that the fluorescence intensity of the recovered amphiphile decreased sharply in the first 10 min, suggesting rapid sequestration of the amphiphile by bacteria, followed by a steady decline in fluorescence intensity that reached a plateau at around 1 h interaction time and remained stable during the entire course of the experiment (up to 6 h). Hence for sake of clarity, the decline in fluorescence intensity of the recovered amphiphile is only shown for a period of 1 h interaction time (Fig. S5, ESI†). In the case of S. aureus at amphiphile concentrations of 2.19, 3.29 and 4.40 μM, the fluorescence intensity of the unbound amphiphile after 10 min interaction time was 94%, 87% and 85%, respectively, in comparison to the compound fluorescence (Fig. S5b, ESI†), indicating dose-dependent binding of compound 6 onto bacterial cell surface, which provides a plausible explanation for the rapid loss of cell viability with increase in amphiphile concentration. A similar trend was observed in the case of E. coli (Fig. S5a, ESI†). Fluorescence microscopic analysis could also track the binding of compound 6 onto bacterial cells and bright fluorescent images of bacteria treated with 4.4 μM of compound 6 could be recorded after 1 h interaction time (Fig. S5c, ESI†).3.5 Mode of action of compound 6
It can be presumed that the cationic nature of compound 6 promotes a multi-target effect and ensures strong binding with the anionic bacterial cell surface. The dose-dependent loss in cell viability with compound 6 (Fig. S4, ESI†) was corroborated by the increase in uptake of PI in amphiphile-treated cells, which provided strong evidence of membrane damage in target bacteria (Fig. 4a and 4b). It can be anticipated that copious membrane damage caused by compound 6 would culminate in loss of cell viability and this fact was ratified by a systematic decrease in cell bound cFDA-SE fluorescence intensities of the cells treated with increasing concentration of compound 6, which clearly indicated a dose-dependent loss in viability of cells (Fig. 4c and 4d). | ||
| Fig. 4 Fluorescence-based assessment of membrane damage and loss in cell viability following interaction with varying concentrations of compound 6. Uptake of PI and cell bound cFDA-SE fluorescence were measured at various time periods in (a and c)E. coli MTCC 433 and (b and d)S. aureus MTCC 96. | ||
Transmission electron microscope (TEM) analysis also provided evidence for membrane damage caused by compound 6. As evident from Fig. 5a control cells of E. coli and S. aureus revealed a characteristic morphology with intact cell wall and prominent electron density within the cells. However, on treatment with 4.4 μM compound 6, significant structural perturbations were observed. Signs of membrane blebbing manifested as irregular surface protrusions were conspicuous in amphiphile-treated cells. Further, there was a marked decrease in intracellular electron density in the amphiphile-treated cells (indicated by an arrow in Fig. 5a) as a consequence of membrane damage and subsequent leakage of intracellular constituents.
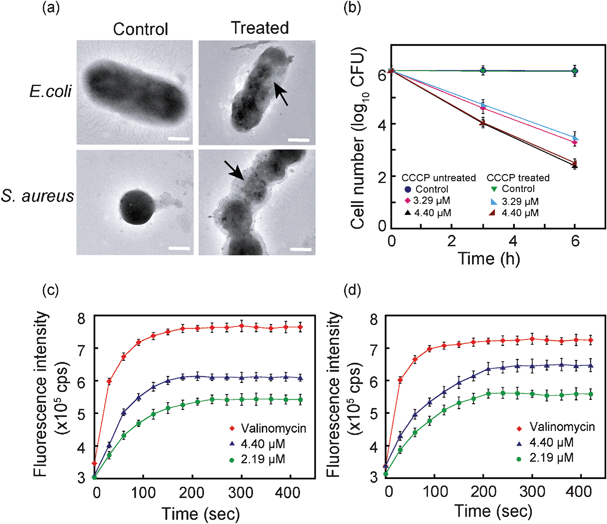 | ||
| Fig. 5 (a) Transmission electron microscopic images of E. coli MTCC 433 and S. aureus MTCC 96. Arrow indicates membrane damage and loss of electron density in cell treated with 4.4 μM compound 6. Scale bar is 0.5 μm. (b) Effect of membrane potential on the bactericidal activity of compound 6 on E. coli MTCC 433. Membrane depolarization assay ascertained by diSC35 fluorescence in (c)E. coli MTCC 433 and (d)S. aureus MTCC 96 cells treated with compound 6. Cells treated with 30 μM valinomycin were used as a positive control for the assay. | ||
The transmembrane potential (ΔΨ) in bacterial cells has been implicated in guiding the activity of antimicrobial agents, such as cationic antimicrobial peptides.11,39 To gain an insight into the mechanism of action of compound 6, cells of E. coli were pre-treated with uncoupler carbonyl cyanide m-chlorophenyl hydrazone (CCCP) to collapse the transmembrane proton motive force and then treated with varying concentrations of compound 6. It was observed that cell viability for CCCP-untreated and CCCP-treated control cells were nearly identical (Fig. 5b), indicating that CCCP treatment per se did not affect cell viability. The viability of cells treated with 4.4 μM compound 6 was less compared to cells treated with 3.29 μM amphiphile for both CCCP-treated and untreated cells, indicating that the difference was solely due to higher dose of amphiphile used for interaction with bacterial cells. Collectively, the results suggested that bactericidal activity of compound 6 was independent of the membrane potential of target bacteria. Similar results have also been reported for the activity of the antimicrobial peptide ranacyclin T on E. coli D21 cells.10 Pathogenic bacterial strains encounter a wide array of host defence mechanisms, which pose a serious impediment to their survival and proliferation. Thus many pathogens have evolved strategies to curb or circumvent these defence mechanisms. Alterations in membrane electrochemistry and energetics has been suggested in case of S. aureus to counter peptide-induced membrane dysfunction and cell death.11 The mode of action of compound 6, which is independent of the characteristic transmembrane potential of bacterial cells, is a distinct advantage and bears significant therapeutic implications, especially in cases where pathogenic bacteria may present an altered membrane electrochemistry.
A correlation between cytoplasmic membrane depolarization and loss in cell viability has been reported for some antimicrobial agents.40,41 To ascertain whether compound 6 could destabilize transmembrane potential in bacterial cells, we used a membrane potential-sensitive probe 3,3′-dipropylthiadicarbocyanine iodide (diSC35). Under the influence of the potential gradient (ΔΨ), the cationic probe accumulates in the cytoplasmic membrane of energized cells, where its fluorescence is quenched. Disruption of the membrane potential gradient leads to release of the probe into solution resulting in an increase in fluorescence intensity.41,42 As seen in Fig. 5c and 5d, treatment of diSC35-loaded E. coli and S. aureus with compound 6 resulted in a rapid increase in fluorescence of the probe, indicating that the amphiphile could dissipate the membrane potential in target cells within a short time. A dose-dependent membrane depolarization effect of compound 6 was also evident. In the case of amphiphile-treated cells, the time taken for diSC35 fluorescence to reach a plateau as well as the magnitude of recovered fluorescence was less compared to the positive control valinomycin, which is a known K+ ionophore. Membrane depolarization by compound 6 was apparently faster in E. coli (Fig. 5c), as the time taken for diSC35 fluorescence to reach a stable value was around 180 s, as compared to 210 s in S. aureus (Fig. 5d). In Gram-negative E. coli, the outer membrane is located just beneath a thin barrier of LPS and is presumably easily accessible to compound 6, whereas in Gram-positive S. aureuscompound 6 has to traverse through a comparatively thick peptidoglycan layer prior to gaining access to the membrane. It is significant to mention that compound 6 could readily cross the LPS barrier and depolarize the membrane in Gram-negative E. coli even without any pre-treatment with membrane-destabilizing agents like EDTA. This property of compound 6 may have important therapeutic implications, as it has been widely acknowledged that LPS plays a pivotal role in enhancing the outer membrane permeability barrier in Gram-negative bacteria and prevents the passage of lipophilic solutes and drugs.43,44
3.6 Cytotoxicity assay
To assess the therapeutic potential of compound 6, it was imperative to determine the cytotoxicity of the amphiphile on human cells. Cytotoxicity of compound 6 on HT-29 cells (human adenocarcinoma cells) was ascertained by XTT colorimetric assay, which measures the activity of mitochondrial dehydrogenase as an index of cell viability. It is evident from Fig. S6 (ESI†) that compound 6 failed to exert any detrimental effect on the metabolic activity of HT-29 cells and there was no decline in the % cell viability in the case of amphiphile-treated cells as compared to the control. Compound 6 was non-cytotoxic to HT-29 cells even at a concentration of 44 μM, which is considerably higher than the concentration required for inhibiting bacterial cell growth. The selective antimicrobial discretion of compound 6 augers well for its therapeutic value and this selectivity may be accounted for by the fundamental difference in membrane phospholipid composition, charge characteristics and energetics between microbial and mammalian cell membranes. The cell membrane in many pathogenic bacteria is electronegative owing to the presence of phosphatidylglycerol (PG), cardiolipin (CL), or phosphatidylserine (PS), as compared to mammalian cytoplasmic membrane, which is rich in zwitterionic phospholipids, such as phosphatidylethanolamine (PE), phosphatidylcholine (PC), sphingomyelin and cholesterol and is thus neutral at physiological pH.11 The electronegative entities present in bacterial cell membranes, such as PG, CL and PS, likely enhance the electro-affinity for cationic compound 6 and probably account for selective antimicrobial targeting of the amphiphile and non-cytotoxicity towards mammalian cells, as reported earlier for other amphiphilic antimicrobial agents.14,45 The results obtained in the present study are encouraging. However, to ratify the non-cytotoxic nature of the compound and to expand the therapeutic application of compound 6, tests on various other cell lines needs to be conducted.4 Conclusions
The present study demonstrates structure-guided tuning of the antimicrobial efficacy of quinoline-based amphiphiles and elucidation of the probable mode of action of the most potent amphiphile. Systematic fluorescence-based assays indicated the dual role of cationic charge and hydrophobic tail length in enhancing the membrane insertion property of the amphiphile, leading to a dramatic increase in antibacterial activity. The most potent amphiphile (compound 6) selected on the basis of structure–activity rationale was highly membrane-active. Experiments suggested that the cationic charge of compound 6 presumably initiates electrostatic interactions, leading to enhanced amphiphile binding on the bacterial cell surface. The hydrophobic tail of the amphiphile steers membrane insertion and sets off membrane dysfunction, probably beginning with collapse of transmembrane potential followed by profound membrane disruption, which ultimately leads to cell death. As a membrane-targeting agent compound 6 may bear significant therapeutic value against drug-resistant pathogens, with advantages such as multi-target effects, rapid and selective bactericidal activity, and a lesser probability of resistance development in target pathogens.The amphiphiles used in the present study provide prototype bio-mimetic scaffolds for generating bactericidal compounds. The structural simplicity of these amphiphiles indicates the possibility of further improvement of antibacterial activity through rational structural design. The method of synthesis of the amphiphile is facile and hence amicable to technological exploitation for mass production. The wide-spectrum bactericidal activity, high potency and lack of cytotoxicity on human cells reflect the therapeutic potential of compound 6. A highlight of the study was the growth inhibition of a presumptive MRSA strain by compound 6 and in future it would be interesting to test the potential of the amphiphile to curb the menace of clinically relevant MRSA and other drug-resistant pathogens. As compound 6 is membrane-active, it would also be worthwhile to surface immobilize the compound in a medical device to prevent bacterial contamination and biofilm formation.
Acknowledgements
We thank the Department of Biotechnology (BT/01/NE/PS/08) and Department of Science and Technology, Government of India for research grants. We also thank Central Instrumental Facility, IIT Guwahati for their valuable help in TEM analysis. M.D.A., B.O. and S.G. acknowledge IIT Guwahati, for a research fellowship. We thank Chockalingam S. for his assistance in cytotoxicity assay.References
- J. M. Thomson and R. A. Bonomo, Curr. Opin. Microbiol., 2005, 8, 518–524 CrossRef CAS.
- M. Morar and G. D. Wright, Annu. Rev. Genet., 2010, 44, 25–51 CrossRef CAS.
- T. D. Gootz, Crit. Rev. Immunol., 2010, 30, 79–93 CAS.
- G. D. Wright, Chem. Commun., 2011, 47, 4055–4061 RSC.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS.
- F. V. Bambeke, M. Leclercq, M. J. Struelens and P. M. Tulkens, Trends Pharmacol. Sci., 2008, 29, 124–134 CrossRef.
- J. G. Hurdle, A. J. O'Neill, I. Chopra and R. E. Lee, Nat. Rev. Microbiol., 2011, 9, 62–75 CrossRef CAS.
- R. E. W. Hancock and D. S. Chapple, Antimicrob. Agents Chemother., 1999, 43, 1317–1323 CAS.
- C. Chen, F. Pan, S. Zhang, J. Hu, M. Cao, J. Wang, H. Xu, X. Zhao and J. R. Lu, Biomacromolecules, 2010, 11, 402–411 CrossRef CAS.
- M. L. Mangoni, N. Papo, G. Mignogna, D. Andreu, Y. Shai, D. Barra and M. Simmaco, Biochemistry, 2003, 42, 14023–14035 CrossRef CAS.
- M. R. Yeaman and N. Y. Yount, Pharmacol. Rev., 2003, 55, 27–55 CrossRef CAS.
- A. K. Marr, W. J. Gooderham and R. E. W. Hancock, Curr. Opin. Pharmacol., 2006, 6, 468–472 CrossRef CAS.
- S. Bera, G. G. Zhanel and F. Schweizer, J. Med. Chem., 2010, 53, 3626–3631 CrossRef CAS.
- S. Dutta, A. Shome and P. K. Das, Langmuir, 2011, 27, 5000–5008 CrossRef CAS.
- E. F. Palermo, I. Sovadinova and K. Kuroda, Biomacromolecules, 2009, 10, 3098–3107 CrossRef CAS.
- E. W. Sugandhi, R. V. Macri, A. A. Williams, B. L. Kite, C. Slebodnick, J. O. Falkinham, A. R. Esker and R. D. Gandour, J. Med. Chem., 2007, 50, 1645–1650 CrossRef CAS.
- I. Baussanne, A. Bussiere, S. Halder, C. Ganem-Elbaz, M. Ouberai, M. Riou, J. M. Paris, E. Ennifar, M. Leclercq and J. L. Decout, J. Med. Chem., 2010, 53, 119–127 CrossRef CAS.
- J. Lv, Y. Qian, T. Liu and Y. Wang, Bioorg. Med. Chem. Lett., 2007, 17, 4102–4106 CrossRef CAS.
- N. A. Lockwood, J. R. Haseman, M. V. Tirrell and K. H. Mayo, Biochem. J., 2004, 378, 93–103 CrossRef CAS.
- K. Kuroda and W. F. DeGrado, J. Am. Chem. Soc., 2005, 127, 4128–4129 CrossRef CAS.
- B. Findlay, G. G. Zhanel and F. Schweizer, Antimicrob. Agents Chemother., 2010, 54, 4049–4058 CrossRef CAS.
- S. R. Meyers, F. S. Juhn, A. P. Griset, N. R. Luman and M. W. Grinstaff, J. Am. Chem. Soc., 2008, 130, 14444–14445 CrossRef CAS.
- B. Ojha and G. Das, Chem. Commun., 2010, 46, 2079–2081 RSC.
- B. Ojha and G. Das, J. Phy. Chem. B., 2010, 114, 3979–3986 CrossRef CAS.
- B. Ojha and G. Das, Chem. Phys. Lipids, 2011, 164, 144–150 CrossRef CAS.
- B. Ojha and G. Das, Photochem. Photobiol. Sci., 2011, 10, 554–560 CAS.
- B. Ojha, A. K. Singh, M. D. Adhikari, A. Ramesh and G. Das, J. Phys. Chem. B, 2010, 114, 10835–10842 CrossRef CAS.
- L. Andree, S. Jean and V. Z. Farchid, Bull. Soc. Chim. Fr., 1987, 6, 1027–1035 Search PubMed.
- C. Weidenmaier and A. Peschel, Nat. Rev. Microbiol., 2008, 6, 276–287 CrossRef CAS.
- T. Gutsmannn and U. Seydel, Eur. J. Cell Biol., 2010, 89, 11–23 CrossRef.
- R. E. Hancock and H. G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS.
- A. Malina and Y. Shai, Biochem. J., 2005, 390, 695–702 CrossRef CAS.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Chem.–Eur. J., 2009, 15, 1123–1133 CrossRef CAS.
- CLSI Performance Standards for Antimicrobial Susceptibility Tests; Seventeenth Informational Supplement. CLSI document M100-S17. Clinical and Laboratory Standards Institute, Wayne, PA, 2007.
- K. S. Kaye and D. Kaye, Curr. Infect. Dis. Rep., 2000, 2, 391–398 CrossRef.
- J. García-Lara, M. Masalha and S. J. Foster, Drug Discovery Today, 2005, 10, 643–651 CrossRef.
- D. Hoefel, W. L. Groobya, P. T. Monisa, S. Andrews and C. P. Saint, J. Microbiol. Methods, 2003, 52, 379–388 CrossRef CAS.
- R. Virto, P. Manas, I. Alvarez, S. Condon and J. Raso, Appl. Environ. Microbiol., 2005, 71, 5022–5028 CrossRef CAS.
- E. Breukink and B. de Kruijff, Biochim. Biophys. Acta, Biomembr., 1999, 1462, 223–234 CrossRef CAS.
- A. Song, S. G. Walker, K. A. Parker and N. S. Sampson, ACS Chem. Biol., 2011, 6, 590–599 CrossRef CAS.
- C. A. Lowery, J. Park, C. Gloeckner, M. M. Meijler, R. S. Mueller, H. I. Boshoff, R. L. Ulrich, C. E. Barry, D. H. Bartlett, V. V. Kravchenko, G. F. Kaufmann and K. D. Janda, J. Am. Chem. Soc., 2009, 131, 14473–14479 CrossRef CAS.
- M. Torrent, S. Navarro, M. Moussaoui, M. V. Nogués and E. Boix, Biochemistry, 2008, 47, 3544–3555 CrossRef CAS.
- R. E. Hancock, Trends Microbiol., 1997, 5, 37–42 CrossRef CAS.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS.
- S. Brahmachari, S. Debnath, S. Dutta and P. K. Das, Beilstein J. Org. Chem., 2010, 6, 859–868 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of amphiphiles, Description of bacterial growth conditions, Screening of antibacterial activity and determination of MIC and MKC, Fluorescence-based structure–function studies, Bactericidal activity of compound 6, amphiphile–bacteria interaction studies, TEM analysis, experimental protocol to study the effect of membrane potential, membrane depolarization assay, data for screening experiment, control experiments, disc-diffusion assay to ascertain methicillin-resistance in S. aureus, time-kill curves, dose-dependent binding of amphiphile on bacterial cells, XTT-based cytotoxicity assay for HT-29 cells, approximate LogP values of neutral amphiphiles, MIC and MKC values. See DOI: 10.1039/c2ra20140b |
| This journal is © The Royal Society of Chemistry 2012 |