Incarviatone A, a structurally unique natural product hybrid with a new carbon skeleton from Incarvillea delavayi, and its absolute configuration via calculated electronic circular dichroic spectra†
Yun-Heng
Shen
a,
Yuanqing
Ding
*b,
Tao
Lu
a,
Xing-Cong
Li
bc,
Jun-Mian
Tian
a,
Hui-Liang
Li
a,
Lei
Shan
a,
Daneel
Ferreira
bc,
Ikhlas A.
Khan
bc and
Wei-Dong
Zhang
*ad
aDepartment of Phytochemistry, School of Pharmacy, Second Military Medical University, 325 Guohe Road, Shanghai 200433, P. R. China. E-mail: wdzhangy@hotmail.com; Fax: +86-21-81871244; Tel: +86-21-81871244
bNational Center for Natural Products Research, Research Institute of Pharmaceutical Sciences, School of Pharmacy, University of Mississippi, MS 38677, USA. E-mail: yqding@olemiss.edu; Fax: +1-662-9157989; Tel: +1-662-9151027
cDepartment of Pharmacognosy, School of Pharmacy, University of Mississippi, MS 38677, USA.
dSchool of Pharmacy, Shanghai Jiao Tong University, Shanghai 200030, P. R. China.
First published on 1st March 2012
Abstract
A phytochemical investigation of Incarvillea delavayi led to the isolation of incarviatone A (1), a structurally unique natural product hybrid with a new carbon skeleton. The structure of 1 was elucidated by spectroscopic analysis, and its absolute configuration was determined by a combination of biogenesis consideration, NOESY experiments, and quantum chemical computational modeling of electronic circular dichroism (ECD).
Introduction
Natural product hybrids are a class of structurally interesting compounds, commonly combining two or more natural products or their fragments to form an unprecedented carbon skeleton.1 By integrating the structural fragments of bioactive substances, natural product hybrids yield not only diverse molecules with unique skeletons, but also potential bioactive therapeutic molecules with novel properties.2 Recently, natural product hybrids have received considerable attention for their diversity and biological activity, and have been considered as new impetus for combinational synthesis and discovering new leads.3 However, because of the structural complexity of this type of compound, it is difficult to determine their structures, especially absolute configurations. Quantum chemical computations, including calculation of electronic circular dichroism (ECD), NMR, and optical rotation (OR), have recently been successfully applied to address this issue, and is increasingly becoming a more powerful tool.4–6In our search for new biologically active therapeutic compounds, we have previously investigated the chemical constituents of several plants of the genus Incarvillea,7–10 which contain structurally diverse monoterpene alkaloids with potent antinociceptive and anti-inflammatory activities.11–14 As a continuation of our investigation on Incarvillea species, incarviatone A (1) (Fig. 1), a structurally unique hybrid comprising a dimeric dehydroiridodial and a benzofuranone-type moiety, was isolated from Incarvillea delavayi, an exquisite mountain flower native to the Yunnan province of China.15 Herein, we report the isolation and structure elucidation of incarviatone A (1). This work also illustrates a successful application of quantum chemical computation in determining the absolute configuration of a non-crystalline complex natural product hybrid.
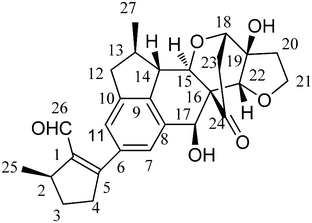 | ||
| Fig. 1 Structure of incarviatone A (1). | ||
Results and discussion
The CHCl3-soluble fraction of the 80% EtOH extract of the whole plant of I. delavayi was subjected to repeated silica gel, RP-18, and Sephadex LH-20 column chromatography to yield compound 1 (4.1 mg).Incarviatone A (1) was isolated as a white amorphous powder. Its UV spectrum showed absorptions at 249 and 273 nm. The positive HR-ESI-MS data indicated a pseudomolecular ion [M+Na]+ at m/z 473.1938, corresponding to a molecular formula C27H30O6 with 13 degrees of unsaturation.
In the 1H NMR spectrum of 1, a formyl proton at δH 9.76 (s), two aromatic protons at δH 7.38 (s) and 7.03 (br.s), four oxygenated methine protons at δH 4.89 (d, J = 10.8 Hz), 4.26 (br.s), 4.24 (d, J = 2.4 Hz), 4.15 (d, J = 9.6 Hz), and one oxymethylene group at δH 4.17 (ddd, J = 1.8, 11.4, 16.2 Hz) and 4.05 (ddd, J = 5.4, 8.4, 16.2 Hz) were observed. The 1H NMR spectrum also indicated the presence of two methyl doublets at δH 1.20 and 1.38 (both d, both J = 6.6 Hz). The 13C NMR and DEPT spectra exhibited 27 carbon resonances, which consisted of two methyls, six methylenes, ten methines, and nine quarternary carbons. Analysis of the 1H and 13C NMR data (Table 1) revealed the presence of several characteristic functionalities, including a formyl group, a tetrasubstituted double bond, a tetrasubstituted benzene ring, a ketone carbonyl group, and two cyclic ether functions. All proton and carbon resonances were assigned by analysis of the 1H and 13C NMR data, together with 2D NMR experiments.
| No. | δ H mult. (J in Hz) | δ C | No. | δ H mult. (J in Hz) | δ C |
|---|---|---|---|---|---|
| a Data were recorded on a Bruker Advance 600 spectrometer; chemical shifts (δ) are given in parts per million with references to the center peak of CDCl3 at δH 7.27 for 1H and δC 77.0 for 13C. | |||||
| 1 | 143.4 | 15α | 4.15 (d, 9.6) | 72.9 | |
| 2α | 3.24 (m) | 39.1 | 16 | 59.2 | |
| 3a | 2.21 (m) | 30.8 | 17α | 4.89 (d, 10.8) | 69.5 |
| 3b | 1.61 (m) | 18α | 4.24 (d, 2.4) | 72.9 | |
| 4a | 3.02 (m) | 37.8 | 19 | 80.4 | |
| 4b | 2.86 (m) | 20a | 2.77 (ddd, 8.4, 11.4, 13.2) | 38.3 | |
| 5 | 163.1 | 20b | 2.00 (ddd, 1.8, 5.4, 13.2) | ||
| 6 | 135.2 | 21a | 4.17 (ddd, 1.8, 11.4, 16.2) | 68.5 | |
| 7 | 7.38 (s) | 124.3 | 21b | 4.05 (ddd, 5.4, 8.4, 16.2) | |
| 8 | 135.1 | 22β | 4.26 (br.s) | 81.7 | |
| 9 | 139.6 | 23a | 2.88 (d, 15.6) | 39.9 | |
| 10 | 142.8 | 23b | 2.73 (dd, 2.4, 15.6) | ||
| 11 | 7.03 (br.s) | 123.6 | 24 | 212.6 | |
| 12a | 2.92 (dd, 7.2, 15.6) | 41.1 | 25 | 1.20 (d, 6.6) | 19.5 |
| 12b | 2.61 (dd, 10.8, 15.6) | 26 | 9.76 (s) | 190.8 | |
| 13α | 2.30 (m) | 44.9 | 27 | 1.38 (d, 6.6) | 19.1 |
| 14β | 2.48 (t, 9.6) | 53.9 | 17-OH | 3.90 (d, 10.8) | |
A 5-methyl-1-formylcyclopentene moiety was established based on the H3-25/H-2/H2-3/H2-4 COSY correlations (Fig. 2), together with the HMBC correlations (Fig. 2) of the formyl proton to C-1 and C-2, and from H3-25 to C-1, C-2, and C-3. Interpretation of COSY data also indicated the presence of the fragment of H-12/H-13(H3-27)/H-14/H-15, which was fused with the benzene ring by HMBC correlations of H-12 with C-11 and C-9, H-14 with C-8 and C-9, and H-17 with C-8. The connectivity of the formyl-substituted methylcyclopentene and benzocyclopentane moieties was confirmed by the observation of HMBC correlations of H-11 and H-7 with C-5, and consequently constituted part a of the structure of 1. Considering the benzofuranone derivatives previously isolated from the genus Incarvillea, in combination with typical signals similar to those of a known compound, cleroindicin F,16 co-existing with compound 1 in the title plant, i.e., a carbonyl group at δC 212.6, an oxymethylene at δC 68.5, an oxymethine at δC 81.7, and an oxygenated quarternary carbon at δC 80.4, showed that 1 contained a cleroindicin F unit (part b). In the HMBC spectrum of 1, the long-range correlations of H-15 with C-18 and C-22, H-17 with C-24 and C-22, H-22 with C-15 and C-17, and H-18 with C-15, strongly supported the linkage of parts a and b as shown. Accordingly, the planar structure of 1 was constructed as shown in Fig. 1.
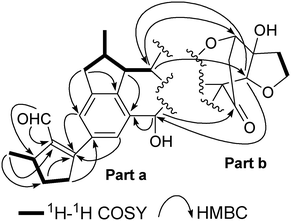 | ||
| Fig. 2 Key 1H–1H COSY and HMBC correlations of incarviatone A (1). | ||
Owing to the unprecedented carbon skeleton and the small quantity of material (ca. 4.1 mg), it was a challenge to determine the stereochemistry of 1. However, consideration of the structural moieties of 1 and its presumed biogenetic pathway could provide some important clues. Monoterpenoid alkaloids and iridoids are biosynthetically derived from iridodial,17–18 and comprise the main constituents of the genus Incarvillea. In addition, we have isolated the known benzofuranone analogue, cleroindicin F, that figures in the structure of 1 as a structural unit. Thus, a plausible biogenetic pathway is proposed as shown in Scheme 1. The key step of this pathway is a Diels–Alder cycloaddition of two dehyroiridodial molecules, and subsequent fusion with a cleroindicin F unit by an aldol condensation reaction to form a structurally unique hybrid. Thus C-19-OH, H-22, and 25-CH3 of 1 should retain their original orientations as shown. In the NOESY spectrum of 1, H-18 was correlated with H-20a, indicating that the bridged ring between C-18 and C-16 should extend above the molecular plane. The NOESY correlation (Fig. 3) of H-14 with H-23b indicated the β-orientation of H-14, with 27-CH3 also β-oriented based on its NOESY correlation with H-14. In addition, the α-orientations of H-15 and H-17 were established by the NOESY correlations from H-13 to H-15 and from H-15 to H-17. Consequently, the relative configuration of 1 was determined as shown in Fig. 1. In order to confirm the above deduction and further determine the absolute configuration of 1, a quantum chemical computational modeling study was conducted by Gaussian 0319 as follows.
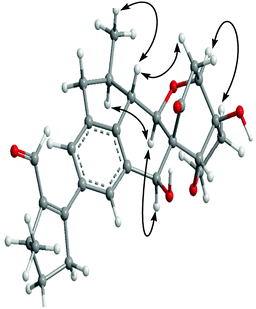 | ||
| Fig. 3 Key NOESY correlations of incarviatone A (1). | ||
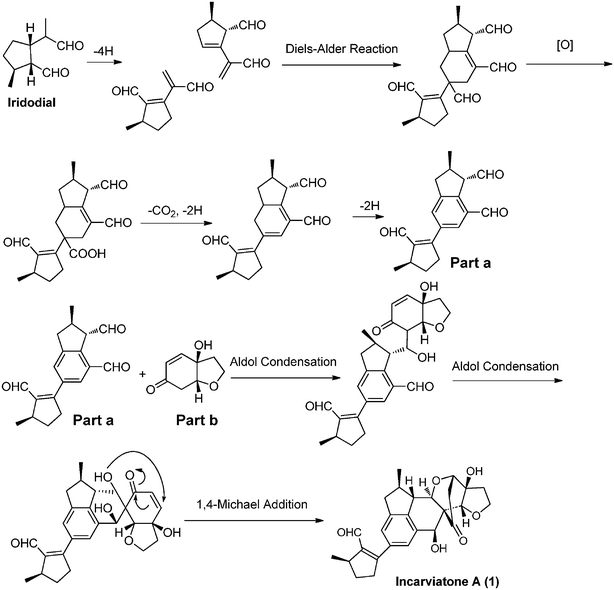 | ||
| Scheme 1 Proposed biogenetic pathway for incarviatone A (1). | ||
The arbitrarily assigned 2R, 13R, 14S, 15R, 16S, 17S, 18S, 19R, 22R absolute configuration of compound 1 has been employed for a systematic conformational random search using an MMFF94 molecular mechanics force-field calculation in the SYBYL 8.1 program (Tripos International, St. Louis, MO). An energy cutoff of 15 kcal mol−1 was used to generate a wide window of conformers in the Boltzmann population, affording 18 conformers. The seven conformers, 1a–1g, within 5 kcal mol−1, were geometrically optimized and relocated in the gas phase using density functional theory (DFT) at the B3LYP/6-31G** level. The major differences among the above seven conformers were the rotations of the C-5–C-6 and C-19–OH bonds. The dihedral angle C-1–C-5–C-6–C-7 for the conformers 1a/1b, 1c/1d, 1e/1f and 1g is 143, −44, −138 and 43°, respectively. The C-19 hydroxy group orients outwards from the cyclic ether in conformers 1a, 1c, 1e and 1g, and inwards in 1b, 1d and 1f, respectively. An additional conformer, 1h, was also accordingly modeled and optimized at the same level. The optimized geometry of 1h is similar to 1g, the dihedral angle C-1–C-5–C-6–C-7 in 1h is 43°, but the C-19–OH group orients inwards. All optimized conformers (1a–1h, Fig. 4) were confirmed as minima by harmonic vibrational frequency computations at the same level. The important optimized dihedral angles are listed in Table S1 (see ESI†). Conformational analysis afforded conformers 1a–1d and 1e–1h with a Boltzman distribution of about 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 (%) at the B3LYP/6-31G** and B3LYP/6-311++G**//B3LYP/6-31G** levels in the gas phase and at the B3LYP-SCRF/6-31G**//B3LYP/6-31G** level with the COSMO model in methanol (Table S2, see ESI†).
:60 (%) at the B3LYP/6-31G** and B3LYP/6-311++G**//B3LYP/6-31G** levels in the gas phase and at the B3LYP-SCRF/6-31G**//B3LYP/6-31G** level with the COSMO model in methanol (Table S2, see ESI†).
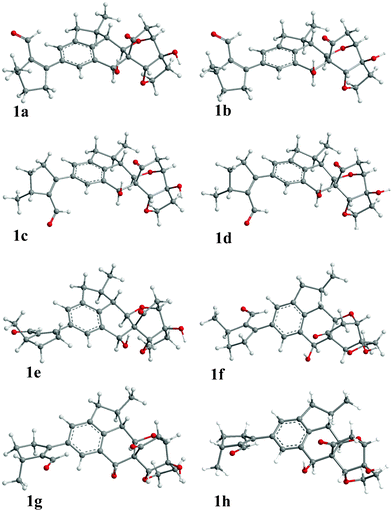 | ||
| Fig. 4 Optimized geometries of predominant conformers of 1 in the gas phase at the B3LYP/6-31G** level. | ||
Time dependent density functional theory (TDDFT) at the above levels were employed to calculate the excitation energy (in nm) and rotatory strength R (velocity form Rvel and length form Rlen in 10−40 erg-esu-cm/Gauss) between different states (Table S4, see ESI†). The electronic circular dichroism (ECD) spectra (Fig. S9, see ESI†) were simulated by overlapping Gaussian functions for each transition.
The overall ECD spectra were weighted by Boltzman distribution (Fig. 5). The observed positive cotton effect (CE) at 351 nm and negative CEs at 296 and 210 nm in experimental ECD spectrum (see Fig. 5 and ESI†) were overall reproduced at about 365, 295 and 211–231 nm. It is noticeable that for conformers 1a–1d (∼40%), the calculated CEs at 365 and 295 nm are opposite to the observed ones but of lower amplitude than those in conformers 1e–1h (∼60%). The calculated ECD curves in conformers 1e–1h agree with the observed spectrum very well. This indicates that the individual ECD spectrum is very conformationally sensitive, especially for the geometrically flexible compounds with multiple free rotated chromophores, and the overall ECD spectrum depends to a high degree on the conformational distribution.
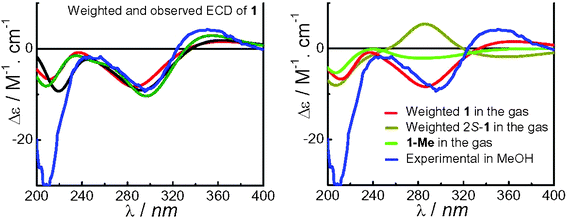 | ||
| Fig. 5 Experimental (blue, in MeOH) and calculated ECD spectra of compound 1 (red, at the B3LYP/6-31G** level in the gas phase: black, at the B3LYP/6-311++G**//B3LYP/6-31G** level in the gas phase; olive, at the B3LYP-SCRF/6-31G**//B3LYP/6-31G** level with the COSMO model in MeOH) and that of 2S-1 (dark yellow) and 1-Me (green) at the B3LYP/6-31G** level in the gas phase. | ||
Molecular orbital analysis of conformer 1e has been performed at the B3LYP/6-31G** level in the gas phase (Table S4, see ESI,†Fig. 6). The major negative rotatory strength at 363 nm and positive rotatory strength at 293 nm are both contributed by the electronic transitions within the formylcyclopentene moiety (MO119→MO121 and MO120→MO121, respectively), the low-amplitute negative rotatory strength at 303 and 285 nm attributed to the n→π* transition of the C-24 carbonyl group (MO117→MO122) and the π→π* transition between the C-24 carbonyl and formyl-cyclopentene moieties (MO120→MO122), respectively. The major negative rotatory strength at 219 nm is produced by transition within the benzene ring. Further calculations confirmed that the above major rotatory strengths at 363 and 293 nm disappeared when the formylcyclopentene moiety was replaced by a methyl group (denoted as 1-Me) while the low-amplitute negative rotatory strength at 303 nm remained (Fig. 5).
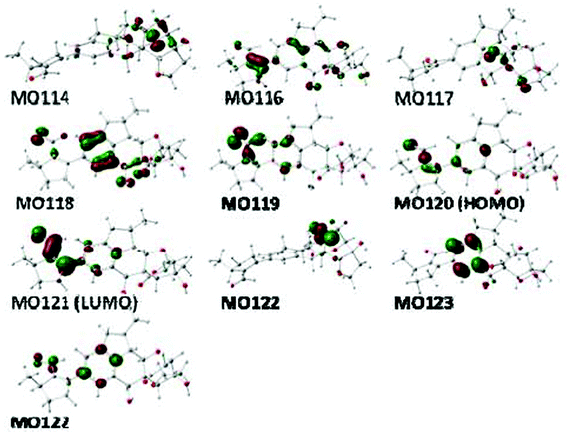 | ||
| Fig. 6 Some molecular orbitals involved in important transitions regarding the ECD spectra of conformer 1e in the gas phase at the B3LYP/6-31G** level. | ||
Since the relative configuration of each stereogenic center, except C-2, in 1 is evidently supported by the NOESY spectrum, their absolute configurations could be either as depicted in 1 or enantiomeric. Thus, it is only necessary to further confirm the absolute configuration at C-2 by reversing it from 2R to 2S (denoted as 2S-1, Fig S10†). Conformational analysis afforded a similar result as for 1, but the predominant conformers are 2S-1a–1d (∼60%, Table S5†) instead of 1e–1h in configuration 1 (Table S2†). The calculated ECD curves of individual conformers of 2S-1 are very similar to that in configuration 1, however, the overall weighted CEs at about 285 and 360 nm were reversed, being opposite to the observed CEs in the same regions (Fig. 5 and Fig. S11†). Therefore, the absolute configuration of 1 could be assigned as 2R,13R,14S,15R,16S,17S,18S,19R,22R.
As a unique hybrid derived from the combination of a dehyroiridodial dimer and a simple benzofuranone analogue, the structure of incarviatone A (1) not only provides insight how nature uilizes seemingly ordinary building blocks to construct elaborate structures with intriguing chemical properties, but also provide an interesting synthetic target.
Experimental section
General experimental procedures
Column chromatography (CC): silica gel (200–300 mesh; Marine Chemical Factory, Qingdao, P. R. China); Sephadex LH-20 (Pharmacia Fine Chemicals, Piscataway, NJ, USA). TLC: silica gel plates, visualization by spraying with 10% H2SO4 in EtOH. Optical rotation: PerkinElmer 343 polar meter. UV spectra: SHIMADAZU UV-2550 spectrophotometer; λmax in nm. NMR Spectra: Bruker DRX-600 (600 MHz) and DRX-400 spectrometer (400 MHz); δ in ppm with SiMe4 as internal standard, J in Hz. MS: Agilent MSD-Trap-XCT (for ESI) and Q-Tof micro mass spectrometer (for HR-ESI), in m/z.Plant material
The whole plants of I. delavayi were collected in Eryuan county, Yunnan province, P. R. China, in July 2006, and authenticated by Prof. Li-Shan Xie of Kunming Institute of Botany, the Chinese Academy of Sciences. A voucher specimen (No. 2006071003) is deposited in School of Pharmacy, Second Military Medical University.Extraction and isolation
The dried whole plants of I. delavayi (17 Kg) were refluxed with 80% EtOH for three times. The combined extract was concentrated to a small volume, and then dissolved in 2% HCl and filtered. The filtrate was adjusted to pH 9-10 by adding 2% NaOH, and then extracted with CHCl3 to give CHCl3 fraction. The CHCl3 fraction (350 g) was subjected to silica gel column chromatography with gradient CHCl3-acetone (100:0–0:100) to afford fractions 1–6. Fraction 2 was purified by repeated column chromatography over reverse phase C18, silica gel and Sephadex LH-20 (CHCl3:MeOH, 1:1) to provide 1 (4.1 mg).
Incarviatone A (1). C27H30O6; White amorphous powder; [α]20D −44.5 (c 0.15, CHCl3); CD (MeOH) Δε210 −30.75, Δε296 −9.29, Δε350 +4.39; UV (MeOH) λmax 249, 273 nm; 1H and 13C NMR data, see Table 1; ESI-MS (negative) 449 [M−H]−, 485 [M+Cl]−; ESI-MS (positive) 473 [M+Na]+; HR-ESI-MS (positive) [M+Na]+m/z 473.1938, calcd for C27H30O6Na, 473.1940.
Computational methods
A systematical conformational random search was conducted by using an MMFF94 molecular mechanics force-field calculation with an energy cutoff of 15 kcal mol−1 in the SYBYL 8.1 program (Tripos International, St. Louis, MO), affording 7 preferred conformers 1a–1g for compound 1. An additional conformer 1h was accordingly modeled based on the geometries of 1a–1g. And so were the conformers 2S-1a–1h for the C-2 epimer of 1. All conformers were geometrically optimized and confirmed as minima by harmonic vibrational frequencies computation in the gas phase using density functional theory (DFT) at the B3LYP/6-31G** level. Time dependent density functional theory (TDDFT) was employed to calculate the excitation energy (in nm) and rotatory strength R (velocity form Rvel and length form Rlen in 10−40 erg-esu-cm/Gauss) between different states at the B3LYP/6-31G** and B3LYP/6-311++G**//B3LYP/6-31G** levels in the gas phase and at the B3LYP-SCRF/6-31G**//B3LYP/6-31G** level with the COSMO model in methanol. The electronic circular dichroism (ECD) spectra were simulated by overlapping Gaussian functions for each transition according to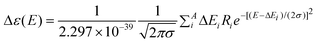 | (1) |
where σ is the width of the band at 1/e height and ΔEi and Ri are the excitation energies and rotatory strengths for transition i, respectively. In this work, σ = 0.20 eV and Rlen were used.
Acknowledgements
The work was supported by program NCET Foundation, NSFC (30725045), partially supported by Global Research Network for Medicinal Plants (GRNMP) and King Saud University, Shanghai Leading Academic Discipline Project (B906), FP7- PEOPLE-IRSES-2008 (TCMCANCER Project 230232), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (10DZ2251300) and the Scientific Foundation of Shanghai China (09DZ1975700, 09DZ1971500, 10DZ1971700). This work was also supported in part by the USDA Agricultural Research Service Specific Cooperative Agreement No. 58-6408-2-0009. We thank the Mississippi Center for Supercomputing Research (MCSR) for computational facilities.References
- K. Gademann, Chimia, 2006, 60, 841–845 CrossRef CAS.
- L. F. Tietze, H. P. Bell and S. Chandrasekhar, Angew. Chem., Int. Ed., 2003, 42, 3996–4028 CrossRef CAS.
- C. Chen, X. Li, C. S. Neumann, M. M.-C. Lo and S. L. Schreiber, Angew. Chem., Int. Ed., 2005, 44, 2249–2252 CrossRef CAS.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914–931 RSC.
- Y. Ding, X. C. Li and D. Ferreira, J. Org. Chem., 2007, 72, 4707–4715 CrossRef.
- P. J. Stephens, J. J. Pan and F. J. Devlin, J. Org. Chem., 2007, 72, 2508–2524 CrossRef CAS.
- T. Lu, W. D. Zhang, Y. H. Pei, C. Zhang, J. C. Li and Y. H. Shen, Chin. Chem. Lett., 2007, 18, 1512–1514 CrossRef CAS.
- Y. C. Chen, Y. H. Shen, Y. Q. Su, L. Y. Kong and W. D. Zhang, Chem. Biodiversity, 2009, 6, 779–783 CAS.
- Y. Q. Su, Y. H. Shen, S. Lin, J. Tang, J. M. Tian, X. H. Liu and W. D. Zhang, Helv. Chim. Acta, 2009, 92, 165–170 CrossRef CAS.
- J. J. Fu, H. Z. Jin, Y. H. Shen, W. D. Zhang, W. Z. Xu, Q. Zeng and S. K. Yan, Helv. Chim. Acta, 2007, 90, 2151–2155 CrossRef CAS.
- Y. M. Chi, F. Hashimoto, W. M. Yan and T. Nohara, Phytochemistry, 1995, 40, 353–354 CrossRef CAS.
- M. Nakamura, Y. M. Chi, W. M. Yan, Y. Nakasugi, T. Yoshizawa, N. Irino, F. Hashimoto, J. Kinjo, T. Nohara and S. Sakurada, J. Nat. Prod., 1999, 62, 1293–1294 CrossRef CAS.
- M. Nakamura, Y. M. Chi, W. M. Yan, A. Yonezawa, Y. Nakasugi, T. Yoshizawa, F. Hashimoto, J. Kinjo, T. Nohara and S. Sakurada, Planta Med., 2001, 67, 114–117 CrossRef CAS.
- M. Nakumura, K. Kido and T. Nohara, Chem. Pharm. Bull., 2000, 48, 1826–1827 CrossRef.
- Editorial committee, Flora of China, Science Press, Beijing, China, 1987, ch. 60, pp. 34–50 Search PubMed.
- J. Tian, Q. S. Zhao, H. J. Zhang, Z. W. Lin and H. D. Sun, Chin. Chem. Lett., 1997, 8, 129–132 CAS.
- M. I. Sampaio-Santos and M. A. C. Kaplan, J. Braz. Chem. Soc., 2001, 12, 144–153 CrossRef CAS.
- T. E. Bellas, W. V. Brown and B. P. Moore, J. Insect Physiol., 1974, 20, 277–280 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, et al., Gaussian 03, Revision D.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1D, 2D NMR spectra, detailed computational data, and ECD data of 1. See DOI: 10.1039/c2ra20188g |
| This journal is © The Royal Society of Chemistry 2012 |