Copper-mediated synthesis of N-fused heterocycles via Csp–S coupling reaction and 5-endo-dig cyclization sequence†
Dongmei
Xiao
a,
Liqiang
Han
a,
Qi
Sun
*a,
Qianxi
Chen
b,
Ningbo
Gong
b,
Yang
Lv
b,
Franck
Suzenet
c,
Gerald
Guillaumet
c,
Tieming
Cheng
a and
Runtao
Li
*a
aState Key Laboratory of Natural and Biomimetic Drugs; School of Pharmaceutical Sciences, Peking University, 38 Xueyuan Road, Beijing, 100191, China. E-mail: sunqi@bjmu.edu.cn; Fax: +86 (10)8271 6956; Tel: +86 (10)8280 1504
bInstitute of Materia Medica (IMM), Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
cInstitut de Chimie Organique et Analytique, Université d'Orléans, UMR-CNRS, 6005, rue de Chartres, BP 6759, 45067 Orléans cedex 2, France
First published on 20th March 2012
Abstract
A novel method was developed for the preparation of N-fused heterocycles via a Csp–S coupling reaction and a sequence of 5-endo-dig cyclization. This method involves the reaction of –NHC(S)NH-containing compounds and alkynes in the presence of CuCl and N,N′-dicyclohexylimidazolium chloride.
Imidazol[2,1-b]-thiazole 1 (Fig. 1a), 5H-thiazolo[3, 2-a]pyrimidine 2 (Fig. 1a) and related N-fused heterocyles represent a class of important heterocyclic compounds that exhibit promising biological activities, such as inhibition of acetylcholinesterase,1 calcium channel antagonistic activity,2 antifungal properties,3 anti-inflammatory properties,4 CDC25 phosphatase inhibitor activity,5 mGluRs antagonist properties,6 and anti-neurodegenerative7 and anti-tumor8 activities. The classical synthetic method for these N-fused heterocycles involves the reaction of 2-mercaptobenzoimidazole 3 or dihydropyrimidine-2-thiones 4 with α-bromo-acetophenones 5 (Fig 1a). This condensation reaction is achieved through –SH alkylation, followed by a dehydration reaction in the presence of a strong acid.9 Although the process is very simple, the reaction is not environmentally friendly, especially when a large amount of α-bromo-acetophenone is used. In addition, extremely acidic reaction conditions will lead to low yields due to hydrolysis.
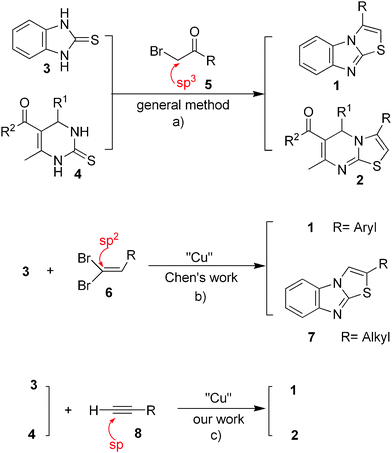 | ||
| Fig. 1 Methods for N-fused heterocycle synthesis. | ||
Transition metal-catalyzed addition of heteroatom-hydrogen bonds to unsaturated organic compounds provides an atom-economic protocol for the construction of C–heteroatom bonds.10 In 2010, Chen11et al. reported a powerful alternative route for the synthesis of heteroatom-containing compounds using sp2-type 1,1-dihaloalkene 6 (Fig 1b), which is considered to be the pre-activated form of the sp3-type α-bromo-acetophenone 5. This catalytic Ullmann-type C-heteroatom coupling is thought to proceed via 1,2-aminothiolation of unsaturated halides, which would yield compounds with N and S nucleophiles at the 1,2-positions. Sequential nucleophilic substitution and H–X (X = S, N) addition would simultaneously result in the formation of C–S and C–N bonds. Although Chen et al. reported that the type of 1,1-dihaloalkene 6 used dictated which of the two isomers 1 and 7 was formed, it is possible that both isomers were simultaneously produced at different ratios, especially for alkyl haloalkenes.
Very recently, Singh et al. developed a one-pot method to synthesize thiazolo[3,2-a]pyrimidine derivatives.12 However, the application of this method, in which bromo-ketones are generated in situ from the reaction of different α-H carbonyl compounds with two equivalents of Br2, may be limited by the requirement for bromine.
Our research group has been engaged in the synthesis of dihydropyrimidine-2-thiones 4 (Fig. 1) and related derivatives for many years.13 These –NHC(S)NH-containing structures have been associated with a broad range of biological activities.14 In particular, they are regarded as calcium channel blockers,15 α-adrenergic antagonists,16 neuropeptide Y antagonists,17 mitotic kinesin Eg5 motor protein inhibitors,18 as well as potent HIV gp-120-CD4 inhibitors.19 In recent years, we have explored the pallado-catalyzed hetero-coupling reaction of 2-thiouracil20 and 421 with tributyltin derivatives in the presence of CuBr·Me2S. In developing an alternative approach to the synthesis of N-fused heterocycles, we envisaged that this transformation could be achieved by a C–S coupling reaction and 5-endo-dig cyclization sequence using inactivated terminal alkynes 8 (Fig. 1c). We herein report a novel route for the synthesis of fused heterocycles 1 and 2via a copper-mediated Csp–S coupling reaction followed by intramolecular hydroamination.
We initially selected the condensation of 2-mercaptobenzo-imidazole 3 and phenylethyne 8a as a model system to optimize the reaction conditions. The results are summarized in Table 1 and show that the types of copper, ligand, base and solvent used were critical to the success of the reaction. The cyclization reaction did not proceed in the absence of CuCl (entry 8, Table 1) or N,N′-dicyclohexylimidazolium chloride (ICy·HCl, entry 6, Table 1). Although similarly good results were obtained with either Et3N (74%, entry 4, Table 1) or NaOBut (72%, entry 1, Table 1), the organic base exhibited advantages during workup. Two equivalents of CuCl and ICy·HCl were used due to the formation of 3–Cu+ complexes and alkynylcopper. Thus, use of fewer equivalents is insufficient, leading to lower yields (entries 2, 3, 5, 7, Table 1). Among the various types of copper screened, CuCl showed the best results (entries 12–15, Table 1). When 0.1 mL of water was added to the reaction system, a much lower yield was obtained (20%, entry 16, Table 1), indicating that moisture was detrimental to the reaction.

| Entry | CuCl (equiv.) | ICy·HCl (equiv.) | Solvent | T (°C) | Time (h) | Yield (%)h |
|---|---|---|---|---|---|---|
| a NaOBut (5 equiv.) was used instead of Et3N. b Reaction conditions: compound 3 (1 mmol), compound 8a (2 mmol), CuCl (198 mg, 2 mmol), ICy·HCl (537 mg, 2 mmol), Et3N (0.71 mL, 5 mmol), in toluene (10 mL). at 110 °C. c 2 equiv. of CuI were used instead of CuCl. d 2 equiv. of CuBr·Me2S were used instead of CuCl. e 2 equiv. of CuTC were used instead of CuCl. f 2 equiv. of CuCl2 were used instead of CuCl. g 0.1 mL H2O was added. h Isolated yields. ICy·HCl = N,N′-dicyclohexyl-imidazolium chloride. | ||||||
| 1a | 2 | 2 | THF | 80 | 48 | 72 |
| 2a | 1 | 1 | THF | 80 | 48 | 40 |
| 3a | 0.5 | 0.5 | THF | 80 | 48 | 32 |
| 4 | 2b | 2 | Toluene | 110 | 24 | 74 |
| 5 | 2 | 1 | Toluene | 110 | 24 | 36 |
| 6 | 2 | — | Toluene | 110 | 24 | Trace |
| 7 | 1 | 2 | Toluene | 110 | 24 | 67 |
| 8 | — | 2 | Toluene | 110 | 24 | Trace |
| 9 | 2 | 2 | Toluene | 110 | 48 | 45 |
| 10 | 2 | 2 | DMF | 110 | 24 | 36 |
| 11 | 2 | 2 | Et3N | 80 | 24 | 40 |
| 12 | 2c | 2 | Toluene | 110 | 24 | 41 |
| 13 | 2d | 2 | Toluene | 110 | 24 | 36 |
| 14 | 2e | 2 | Toluene | 110 | 24 | Trace |
| 15 | 2f | 2 | Toluene | 110 | 24 | 32 |
| 16 | 2g | 2 | Toluene | 110 | 24 | 20 |
The scope of the study was then expanded to examine the effect of different aromatic terminal alkynes (8b–8f) on the copper-mediated synthesis of N-fused heterocyclic compounds using 2-mercaptobenzoimidazole 3 and the optimized protocol identified in Table 1 (entry 4). As shown in Table 2, use of the electron-rich (8b and 8c) and electron-poor (8d) aromatic terminal alkynes provided the corresponding products in good yields (entries 2–4, Table 2). Different heteroaryl alkynes afforded the desired products 1e and 1f in 63% and 41% yields, respectively (entries 5 and 6, Table 2).

Next, the synthesis of N-fused heterocycles from the reaction of phenylethyne 8a with different dihydropyrimidine-2-thiones 4a–n was investigated, as shown in Table 3. The results show that the reaction can tolerate various substituents on the 4-phenyl. Dihydropyrimidine-2-thiones with m-substituted phenyl groups bearing a halogen or electron-donating group afforded the desired products with excellent yields (entries 2–5, Table 3). Dihydropyrimidine-2-thiones with an m-nitrophenyl group 4f, as well as those with p-substituted phenyl groups (4g–i) gave products with good yields (entries 6–9, Table 3). In contrast, only moderate yields were isolated from dihydropyrimidine-2-thiones with o-substituted phenyl groups (entries 10–12, Table 3), suggesting that steric effects from the phenyl substitutents influenced the reaction. Replacing the 5-ester group with a keto group did not reduce the yield (entry 13, Table 3). A heteroaryl substituent afforded the desired product 4n in 63% yield (entry 14).

| Entry | R1 | R2 | Product | Yield (%)b |
|---|---|---|---|---|
| a Reactions were run on a 1 mmol scale of 4a–n with 2 mmol of 8a using the optimized protocol. b Isolated yields. | ||||
| 1 | Ph | OMe (4a) | 2a | 75 |
| 2 | 3-F-Ph | OMe (4b) | 2b | 98 |
| 3 | 3-Cl-Ph | OMe (4c) | 2c | 93 |
| 4 | 3-Br-Ph | OMe (4d) | 2d | 88 |
| 5 | 3-OMe-Ph | OMe (4e) | 2e | 89 |
| 6 | 3-NO2-Ph | OMe (4f) | 2f | 72 |
| 7 | 4-OMe-Ph | OMe (4g) | 2g | 68 |
| 8 | 4-Me-Ph | OMe (4h) | 2h | 82 |
| 9 | 3,4-OCH2O-Ph | OMe (4i) | 2i | 75 |
| 10 | 2-OMe-Ph | OMe (4j) | 2j | 67 |
| 11 | 2-Br-Ph | OMe (4k) | 2k | 66 |
| 12 | 2-Cl-Ph | OMe (4l) | 2l | 67 |
| 13 | Ph | Me (4m) | 2m | 83 |
| 14 | 2-Thiophenyl | OMe (4n) | 2n | 63 |
Reactions of dihydropyrimidine-2-thione 4c with different aromatic (8b–8d) and aliphatic (8g and 8h) terminal alkynes are summarized in Table 4. As shown, electron-rich (8b) and electron-poor (8d) aromatic terminal alkynes provided the corresponding products in good yields (entries 1 and 3, Table 4). The use of aliphatic terminal alkynes 8g and 8h were also shown to be feasible and resulted in products 2r and 2s with 67% and 68% yields (entries 4–5, Table 4).

In order to detemine the structure of the isolated 5H-thiazolo[3, 2-a]pyrimidine, we compared the 1H NMR, 13C NMR and HRMS spectra of the products with literature reports.1 In addition, we also analyzed the crystal structure of compound 2a using X-ray diffraction measurement (Fig. 2).
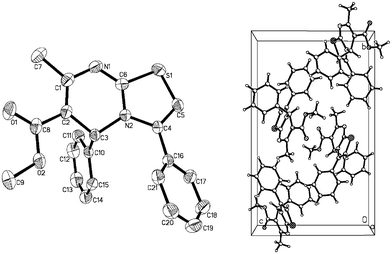 | ||
| Fig. 2 X-ray crystal structure of compound 2a. | ||
Based on the results, we propose the reaction mechanism illustrated in Scheme 1. Under the standard reaction conditions, the precursor 9 was easily formed in accordance with Tatibouët's work.22 The oxidative coupling reaction of 9 and 3 affords the key intermediate 10, which is then transformed into the alkynyl structure 11via transmetalation. Subsequently, the N-fused heterocyclic compound 12 is formed via 5-endo-dig cyclization, followed by Cu+–H+ exchange to give the final N-fused heterocycle 1a.
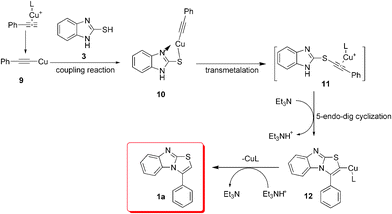 | ||
| Scheme 1 Proposed mechanism for copper(I)-mediated N-fused heterocycle formation. | ||
In conclusion, we have developed a novel method for the synthesis of N-fused heterocycles via Csp–S coupling and a sequence of 5-endo-dig cyclization. This procedure has been demonstrated to be economical, simple and facile for the preparation of N-fused heterocyclic derivatives from –NHC(S)NH-containing compounds and terminal alkynes in the presence of CuCl and ICy·HCl. The structures of dihydropyrimidine derivatives were confirmed by X-ray analysis of compound 2a. Moreover, a possible mechanism is proposed for this novel reaction.
The project is supported by NSFC (No. 20802004). We thank Professor Ning Jiao (Peking University) for his review and comments. We also thank Dr Samuel K. Kulp (The Ohio State University) for his help with improving the English grammar.
References
- H. Zhi, L. Chen, L. Zhang, S. Liu, Z. Wen, H. Lin and C. Hu, Chin. J. Med. Chem., 2008, 18, 340 CAS.
- A. Balkan, S. Uma, M. Ertan and W. Wiegrebe, Pharmazie, 1992, 47, 687 CAS.
- M. M. Ghorab, Y. A. Mohamad, S. A. Mohamed and Y. A. Ammar, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 108, 249 CrossRef CAS.
- B. Tozkoparan, M. Ertan, P. Kelicen and R. Demirdamar, Farmaco, 1999, 54, 588 CrossRef CAS.
- R. Duval, S. Kolb, E. Braud, D. Genest and C. Garbay, J. Comb. Chem., 2009, 11, 947 CrossRef CAS.
- J. Wichmann, G. Adam, S. Kolczewski, V. Mutel and T. Woltering, Bioorg. Med. Chem. Lett., 1999, 9, 1573 CrossRef CAS.
- N. Pietrancosta, A. Moumen, R. Dono, P. Lingor, V. Planchamp, F. Lamballe, M. Bähr, J.-L. Kraus and F. Maina, J. Med. Chem., 2006, 49, 3645 CrossRef CAS.
- T. U. Mayer, T. M. Kapoor, S. J. Haggarty, R. W. King, S. L. Schreiber and T. J. Mitchison, Science, 1999, 286, 971 CrossRef CAS.
- (a) H. F. Andrew and C. K. Bradsher, J. Heterocycl. Chem., 1967, 4, 577 CrossRef CAS; (b) S. A. T. Narayan, V. Kumar and H. K. Pujari, Indian J. Chem., Sec B: Org. Chem. Incl. Med. Chem., 1986, 25B(3), 267 CAS.
- (a) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef; (b) J. Han, B. Xu and G. B. Hammond, J. Am. Chem. Soc., 2010, 132, 916 CrossRef CAS; (c) J. Yang, A. Sabarre, L. R. Fraser, B. O. Patrick and J. A. Love, J. Org. Chem., 2009, 74, 182 CrossRef CAS; (d) A. Sabarre and J. Love, Org. Lett., 2008, 10, 3941 CrossRef CAS.
- H. Xu, Y. Zhang, J. Huang and W. Chen, Org. Lett., 2010, 12, 3704 CrossRef CAS.
- S. Singh, A. Schober, M. Gebinoga and G. A. Groβ, Tetrahedron Lett., 2011, 52, 3814 CrossRef CAS.
- Q. Sun, Y. Q. Wang, Z. M. Ge, T. M. Cheng and R. T. Li, Synthesis, 2004, 7, 1047 Search PubMed.
- C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS.
- H. Cho, M. Ueda, K. Shima, A. Mizuno, M. Hayashimatsu, Y. Ohnaka, Y. Takeuchi, M. Hamaguchi and K. Aisaka, J. Med. Chem., 1989, 32, 2399 CrossRef CAS.
- (a) D. Nagarathnam, S. W. Miao, B. Lagu, G. Chiu, J. Fang, T. G. M. Dhar, J. Zhang, S. Tyagarajan, M. R. Marzabadi, F. Q. Zhang, W. C. Wong, W. Y. Sun, D. Tian, J. M. Wetzel, C. Forray, R. S. L. Chang, T. P. Broten, R. W. Ransom, T. W. Schorn, T. B. Chen, S. O'Malley, P. Kling, K. Schneck, R. Benedesky, C. M. Harrell, K. P. Vyas and C. Gluchowski, J. Med. Chem., 1999, 42, 4764 CrossRef CAS; (b) J. C. Barrow, P. G. Nantermet, H. G. Selnick, K. L. Glass, K. E. Rittle, K. F. Gilbert, T. G. Steele, C. F. Homnick, R. M. Freidinger, R. W. Ransom, P. Kling, D. Reiss, T. P. Broten, T. W. Schorn, R. S. L. Chang, S. S. O'Malley, T. V. Olah, J. D. Ellis, A. Barrish, K. Kassahun, P. Leppert, D. Nagarathnam and C. Forray, J. Med. Chem., 2000, 43, 2703 CrossRef CAS.
- M. A. Bruce, G. S. Poindexter and G. Johnson, 1999, US 5889016 A 19990330 Search PubMed.
- S. J. Haggarty, T. U. Mayer, D. T. Miyamoto, R. Fathi, R. W. King, T. J. Mitchison and S. L. Schreiber, Chem. Biol., 2000, 7, 275 CrossRef CAS.
- A. D. Patil, N. V. Kumar, W. C. Kokke, M. F. Bean, A. J. Freyer, C. De Brosse, S. Mai, A. Truneh, D. J. Faulkner, B. Carte, A. L. Breen, R. P. Hertzberg, R. K. Johnson, J. W. Westley and B. C. M. Potts, J. Org. Chem., 1995, 6, 1182 CrossRef.
- Q. Sun, F. Suzenet and G. Guillaumet, J. Org. Chem., 2010, 75(10), 3473 CrossRef CAS.
- Q. Sun, F. Suzenet and G. Guillaumet, Tetrahedron Lett., 2012 DOI:10/1016/j.tetlet.2012.03.067.
- S. Silva, B. Sylla, F. Suzenet, A. Tatibouët, A. P. Rauter and P. Rollin, Org. Lett., 2008, 10, 853 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental procedures, spectroscopic data, crystallographic data of 2a in CIF format. See DOI: 10.1039/c2ra20254a |
| This journal is © The Royal Society of Chemistry 2012 |