Aromatization of IMDAF adducts in aqueous alkaline media†
Fedor I.
Zubkov
*,
Inga K.
Airiyan
,
Julya D.
Ershova
,
Timur R.
Galeev
,
Vladimir P.
Zaytsev
,
Eugeniya V.
Nikitina
and
Alexey V.
Varlamov
Department of Organic Chemistry, Peoples' Friendship University of Russia, 6 Miklukho-Maklaya St., Moscow, Russia. E-mail: fzubkov@sci.pfu.edu.ru
First published on 21st February 2012
Abstract
In this paper, we propose a simple synthesis of isoindoline-4-carboxylic acids by means of the aromatization of 3a,6-epoxyisoindoles in alkaline media. The method is facile from an experimental point of view: a short-term (0.5–2h) reflux of epoxyisoindoles in 5% aqueous solutions of alkali leads to the target products in 40–90% yields. The absence of by-products, ease of isolation of the target products and applicability to acidophobic group bearing substrates favorably distinguishes the proposed procedure from previously utilized acid-catalyzed methods. The proposed strategy has been successfully utilized for isoindole containing compounds and nuevamine-type alkaloids.
Introduction
Isoindole chemistry is a thriving and relatively new area of organic chemistry; nevertheless, numerous synthetic approaches toward isoindoles have been proposed in the literature.1 This advance is due to the large amount of biologically active isoindole-containing compounds found in nature2 (for example, lennoxamine, aristoyagonine, nuevamine, chilenine and some other alkaloids).Of all the isoindole fragment construction methods, the one that has attracted the most attention of our group over the last decade3 is a two-step approach based on the IMDAF (intramolecular Diels–Alder reaction of furans)4 reaction. The advantages of the method are cheap starting materials, wide chemical diversity (substituted furfurals can be used as the starting materials and various alkenyl halides, anhydrides and chlorides of α,β-unsaturated acids can be used in the acylation step) and the simplicity of the experimental procedure.
We have been trying to apply this approach for syntheses of isoindole-containing alkaloids and alkaloid-like compounds, particularly nuevamine and its derivatives (Fig. 1). We encountered problems in the final step of the route for our model compounds (see Results and discussion): aromatization of the 7-oxabicyclo[2.2.1]heptene fragment to 10,12a-epoxyisoindolo[1,2-a]isoquinoline using acid catalysis and common procedures was not successful.
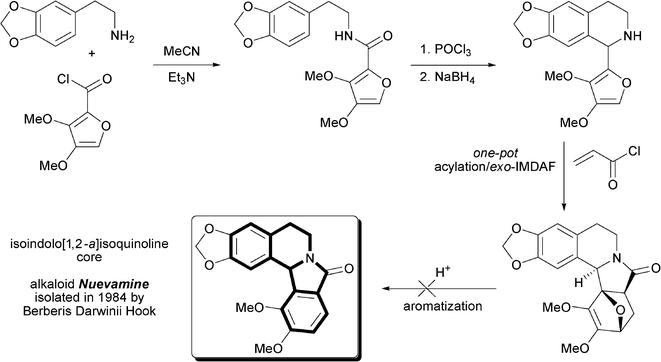 | ||
| Fig. 1 Proposed synthesis of nuevamine via the IMDAF reaction | ||
The IMDAF approach towards isoindolines (Fig. 2) was suggested for the first time by Yugoslav (now Croatian) chemists in 1964.5 The authors proposed a number of reagents to be utilized in the final step as dehydrating agents: HBr/AcOH, H2SO4/AcOH and diluted H2SO4. This approach turned out to be so effective that it is still being widely used6 and improved.7 Thereupon, the method was extended to aromatization of the 3a,6-epoxy-2-benzofuran core.8
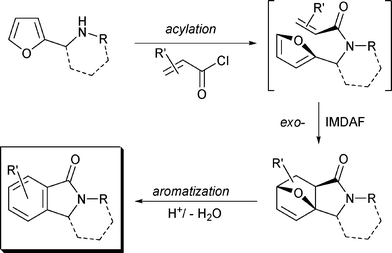 | ||
| Fig. 2 The key step-aromatization of 3a,6-epoxyisoindole fragment | ||
In addition to the above-mentioned reagents, H3PO4,3 HCl,6ap-TSA6g,7a and BF3·OEt29 are also effective tools for this transformation. Two more methods7 of aromatization of 3a,6-epoxyisoindoles have been proposed: a protic ionic liquid based approach7b (CF3SO3H/ionic liquid/microwave irradiation), and a method utilizing the K10-Fe3+ catalyst coated on montmorillonite clay.7c
Most of the mentioned conditions lead to good yields of the target isoindoles. However, all cited methods involve acid catalysis. It should be noted that proton or Lewis acids can produce unpredictable but often very interesting side transformations of the 7-oxabicyclo[2.2.1]heptene fragment.5f,6e,9 See for example, the unexpected rearrangement of 2,3,7,7a-tetrahydro-3a,6-epoxyisoindol-1-ones to furo[2,3-c]pyrrolo[3,4-b]pyrrole-2,4,6-triones in 85% H3PO4.9 Another limitation of the described methods is their nonapplicability to the synthesis of isoindoles bearing acidophobic substituents (furyl, allyl, pyrrolyl, etc.)
There are a few studies on the aromatization of the 7-oxabicyclo[2.2.1]heptene moiety in strong alkaline media. As an illustration, a very limited number of publications on the aromatization of similar systems with metal–organic compounds and alcoholates can be mentioned.8a,10 Dehydrative aromatization of 3-oxo-1,3-dihydro-2-benzofuran-4-carboxylic acids takes place in a 3M NaOMe solution8a and aromatization of 7-oxabicyclo[2.2.1]heptanes can be carried out using t-BuLi.10a Aromatization of epoxyisoindolyl phosphonates10b can be performed in different conditions (H3PO4, TFA), but NaOMe/MeOH leads to several by-products.
We present a newly developed effective procedure of aromatization of 3a,6-epoxyisoindoles in 5% alkali aqueous solution, which was successfully utilized for the synthesis of nuevamine derivatives. To the best of our knowledge, this is the first time dilute aqueous alkaline solutions have been used as reaction media for this transformation. The approach allows the use of the Diels–Alder adducts of furan for the synthesis of diverse heterocycles annelated with a benzene ring, including ones bearing acidophobic fragments.
Results and discussion
The starting materials, exo-1-oxo-1,2,3,6,7,7a-hexahydro-3a,6-epoxyisoindole-7-carboxylic acids 2a–l, were easily prepared from the commercially available amines and furfural in two steps (Scheme 1).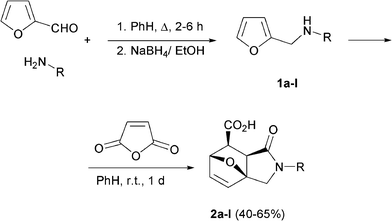 | ||
| Scheme 1 Synthesis of the prerequisite 3,6a-epoxyisoindoles. | ||
The reaction was performed according to the typical procedure.3d,7a Furfurylamines 1a–l were purified by vacuum distillation and the target adducts 2a–l were obtained in a total yield of 40–65% (overall yield relative to furfural).
In order to study reactivity of the synthesized lactams, we tried to carry out the hydrolysis of the amide fragment of 2a in aqueous alkaline media. To our surprise, treatment of adduct 2a with a 10% aqueous solution of sodium hydroxide did not lead to 1-(aminomethyl)-7-oxabicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid (Scheme 2).
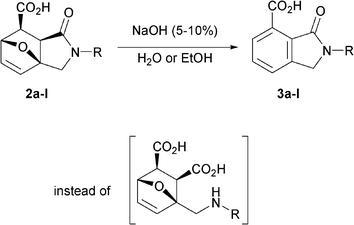 | ||
| Scheme 2 Aromatization of 3a,6-epoxyisoindole-7-carboxylic acids 2a–l in alkaline medium. | ||
Instead, 3-oxoisoindoline-4-carboxylic acid 3a was obtained as a single product in good yield. Then we optimized the experimental conditions to achieve maximum yields of the acids, 3, (Table 1) with the intention of developing a robust method of aromatization of 3a,6-epoxyisoindoles in aqueous alkaline media, which could be used as an alternative to the methods involving acidic conditions (Scheme 1).
| Comp. | Base | Conc. (%) | Solvent | Time (h) | Yield (%) | |
|---|---|---|---|---|---|---|
| before | aftera | |||||
| recrystallization | ||||||
| a Recrystallization from an i-PrOH/DMF mixture. | ||||||
| 3a | NH3 | 25 | H2O | 2 | 0 | 0 |
| 3a | NaOH | 2 | H2O | 2 | 0 | 0 |
| 3a | NaOH | 5 | H2O | 0.5 | 92 | 52 |
| 3a | NaOH | 5 | H2O | 1 | 86 | 53 |
| 3a | NaOH | 5 | H2O | 2 | 70 | 49 |
| 3a | NaOH | 10 | H2O | 1.5 | 67 | 50 |
| 3a | NaOH | 5 | EtOH | 0.5 | 90 | 43 |
| 3b | NaOH | 5 | EtOH | 2 | 83 | 45 |
| 3b | NaOH | 5 | MeOH | 2 | 0 | 0 |
| 3b | NaOH | 10 | H2O | 2 | 75 | 41 |
| 3b | NaOH | 10 | EtOH | 1 | 89 | 46 |
| 3b | NaOH | 15 | EtOH | 1 | 82 | 63 |
The most accessible epoxides 2a,b were used as model substrates to optimize the yield. The reaction does not take place in 2–3% alkali (water or EtOH solutions) or a concentrated solution of ammonia (Table 1). The best results were obtained with 5–10% alkali solutions in water or ethanol (the yields are comparable). Reflux duration depends slightly on the structure of the initial substrate (the starting acid must be completely dissolved) and does not exceed 1.5 h. Apparently, the reaction temperature is required to be higher than 70 °C, since aromatization did not take place when the mixture was refluxed in a 5% NaOH methanol solution. Thus, for all further syntheses (see Scheme 2 and Table 2), we used a 10% solution of NaOH in water, with a 1.5 h reflux time.
| Compounda | R | Empirical formula | Found (%) | m.p. (°C)b | IR (KBr) | Yieldb (%) | ||
|---|---|---|---|---|---|---|---|---|
| Calculated (%) | ν (cm−1) | |||||||
| C | H | N | CO2H/NCO | |||||
| a All compounds were obtained under the following conditions: a solution of NaOH (2.2 g, 56 mmol) and 1 g (3 mmol) of 2, 5 or 7 in 20 mL of water was refluxed for 1.5 h. See the Experimental section for the work-up procedure. b Yields and melting points of all compounds are given after recrystallization from i-PrOH/DMF. c Yields of the individual diastereoisomers of 8 are given after fractional crystallization, the total yields of 8a and 8b are given in the Experimental section. | ||||||||
| 3a | Ph | C15H11NO3 | 71.32 | 4.54 | 5.71 | 228.9–229.6 | 1714/1610 | 49 |
| 71.14 | 4.38 | 5.53 | ||||||
| 3b | Bn | C16H13NO3 | 71.71 | 4.87 | 5.54 | 183–185 | 1713/1600 | 43 |
| 71.90 | 4.90 | 5.24 | ||||||
| 3c | Me | C10H9NO3 | 62.42 | 4.34 | 7.15 | 207.6–210.9 | 1704/1609 | 65 |
| 62.82 | 4.74 | 7.33 | ||||||
| 3d | Et | C11H11NO3 | 64.78 | 5.21 | 6.56 | 195.9–196.7 | 1712/1608 | 60 |
| 64.38 | 5.40 | 6.83 | ||||||
| 3e | Allyl | C12H11NO3 | 66.65 | 5.31 | 6.75 | 166.7–168.2 | 1710/1608 | 45 |
| 66.35 | 5.10 | 6.45 | ||||||
| 3f | Furfuryl | C14H11NO4 | 65.27 | 4.11 | 5.74 | 231.6–234.8 | 1706/1590 | 53 |
| 65.37 | 4.31 | 5.44 | ||||||
| 3g | –(CH2)4CH3 | C14H17NO3 | 68.34 | 6.72 | 5.36 | 144.4–144.9 | 1711/1609 | 47 |
| 68.00 | 6.93 | 5.66 | ||||||
| 3h | –CH2C6H3Cl2-2,3 | C16H11Cl2NO3 | 57.41 | 3.16 | 4.51 | 223–226 | 1704/1605 | 35 |
| 57.16 | 3.30 | 4.17 | ||||||
| 3i | (CH2)2C6H3(OMe)2-3,4 | C19H19NO5 | 66.65 | 5.81 | 4.41 | 193–194 | 1705/1605 | 48 |
| 66.85 | 5.61 | 4.10 | ||||||
| 3j | 3-Cl,4-Me-C6H3 | C16H12ClNO3 | 63.39 | 4.22 | 4.85 | 259–262 | 1719/1597 | 30 |
| 63.69 | 4.01 | 4.64 | ||||||
| 3k | Cyclohexyl | C15H17NO3 | 69.26 | 6.98 | 5.12 | 245–247 | 1703/1590 | 46 |
| 69.48 | 6.61 | 5.40 | ||||||
| 3l | 5-Methyl-1,2-oxazol-3-yl | C13H10N2O4 | 60.35 | 3.84 | 11.05 | 271.8–272.5 | 1726/1634 | 38 |
| 60.47 | 3.90 | 10.85 | ||||||
| 6a | H | C17H13NO3 | 73.31 | 4.52 | 5.24 | 194–195 (decomp.) | 1716/1610 | 60 |
| 73.11 | 4.69 | 5.02 | ||||||
| 6b | OMe | C19H17NO5 | 67.33 | 5.20 | 4.30 | 160–161 (decomp.) | 1732/1615 | 55 |
| 67.25 | 5.05 | 4.13 | ||||||
| 8aA | — | C19H15NO4 | 70.10 | 4.76 | 5.03 | 236–238 | 1705/1620 | 7c |
| 71.02 | 4.71 | 4.36 | ||||||
| 8aB | — | C19H15NO4 | 70.91 | 4.50 | 4.70 | 215–217 | 1722/1625 | 56c |
| 71.02 | 4.71 | 4.36 | ||||||
| 8bA | — | C20H17NO4 | 71.58 | 5.39 | 4.49 | 259.6–260.6 | 1706/1623 | 14c |
| 71.63 | 5.11 | 4.18 | ||||||
| 8bB | — | C20H17NO4 | 71.52 | 5.43 | 4.30 | 235–237 | 1713/1622 | 27c |
| 71.63 | 5.11 | 4.18 | ||||||
The exceptional ease of isolating the final products, 3, should be stressed. No extraction with organic solvents or column chromatography purification is required. After neutralization (diluted H2SO4) pure (according NMR 1H data) target products 3 are separated by simple filtration and the only by-product is sodium sulfate.
Even after recrystallization (which in general leads to a 20–30% loss in yield—see Table 1) the yields of isoindolinecarboxylic acids 3 (Table 2) are comparable to those described in the literature.3d,5,7a,7b,10b Recrystallization was performed in all cases in order to obtain analytically pure samples.
With the alkaline aromatization methodology developed for simple model systems, we proceeded to more complicated structures. The leitmotif of the further study was the investigation of the scope and limitations of the method applied to 3a,6-epoxyisoindoles condensed with other heterocycles (Schemes 3 and 4). A large number of such structures are described in the literature.3,4
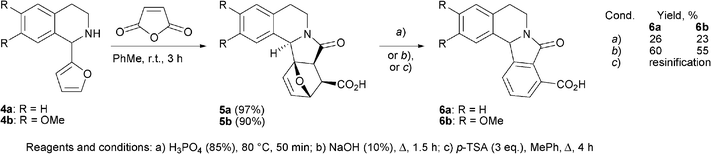 | ||
| Scheme 3 Test synthesis of the Nuevamine-like systems. | ||
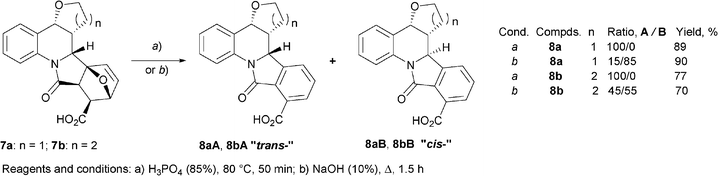 | ||
| Scheme 4 Observed epimerization in the course of alkaline aromatization. | ||
The advantage of the procedure can be illustrated by the aromatization of the model nuevamine-like structures 511 containing the isoindolo[1,2-a]isoquinoline skeleton (Scheme 3 and Table 2). A comparison of acidic and basic reaction conditions for the synthesis of 6 shows that the latter lead to significantly higher yields of 6 and, apparently, may be successfully used in total synthesis of nuevamine.
The alkaline medium may lead to epimerization of the final products (Scheme 4 and Table 2). Aromatization of furo- or pyrano[3,2-c]-annelated 6b,9-epoxyisoindolo[1,2-a]quinolines 712 in basic conditions leads to a mixture of diastereomers 8A/8B, while the epimerization was not observed in the acidic conditions. In an independent experiment it was established that the isomerization of 8A to 8B occurs in the same conditions. The 8A/8B ratio in the formed mixtures is approximately the same.
Conclusions
We present an experimentally facile two-step method for the synthesis of isoindolone derivatives, which is based on the cleavage of IMDAF adducts in alkaline media. The availability of starting materials, mild conditions, benign reaction media and simple isolation procedure involving simple filtration make this approach very attractive. The method is a more robust alternative to the commonly used aromatization procedures in acidic conditions. Also, it makes previously inaccessible isoindoles bearing acidophobic substituents available and is preferential in cases leading to undesirable rearrangements under acidic conditions. We demonstrated the advantage of the developed aqueous alkaline medium procedure over the commonly used acidic conditions methods by the synthesis of nuevamine-like systems.Experimental
All reagents were purchased from Acros Chemical Co. All solvents were used without further purification. Melting points were determined using a SMP30 and are corrected. IR spectra were obtained in KBr pellets for solids using an IR-Fourier spectrometer Infralum FT-801. NMR spectra 1H (400 or 600 MHz) and 13C (100.6 or 150.9 MHz) were recorded for solutions in deuteriochloroform or DMSO-d6 at 27 °C and traces of chloroform (1H NMR δ 7.26 ppm and 13C NMR 76.90 ppm) or DMSO-d5H (1H NMR δ 2.49 ppm and 13C NMR 39.43 ppm) were used as the internal standard. Mass spectra were measured either on Thermo Trace DSQ (electron ionization, 70 eV, ion source temperature was 200 °C, direct inlet probe) or on JEOL AccuTOF JMS-T100LP spectrometer using direct analysis in real time (DART) ionization method (helium was used as DART gas, gas flow rate was 1 L min−1, flow T 300 °C, discharge electrode was set to +4000 V, the mass scale was calibrated using PEG 600). The purity of the obtained substances and the composition of the reaction mixtures were controlled by TLC Sorbfile plates (using an EtOH/AcOH mixture as eluent). The separation of the final products was carried out by the crystallization from i-PrOH/DMF. Microanalysis was performed for C, H, N on a Vario Macro Cube C,H,N,O,S-analyser and were within ± 0.35% of theoretical values.Hexahydro-3a,6-epoxyisoindol-7-carboxylic acids 2; General Procedure
A mixture of maleic anhydride (9.8 g, 0.1 mol) and N-R-furfurylamine 1a–k (0.1 mol) in benzene (100 ml) was stirred at 25 °C for 24 h. The precipitate formed was filtered off, washed with benzene (50 mL), ether (50 mL) and dried at 85 °C to constant weight. Compounds 2a–l were obtained in the form of fine crystalline white powders, which did not require further purification (satisfactory results of elemental analysis and NMR spectra).Representative characteristics of epoxydes 2a,2b (all other data for 2c–2l are given in the ESI†) (3aS*,6R*,7S*,7aR*)-1-Oxo-2-phenyl-1,2,3,6,7,7a-hexahydro-3a,6-epoxyisoindol-7-carboxylic acid (2a)
Yield: 86%; m.p. 183.1–184.9 °C.
This compound was obtained and described previously3d,5b [lit.3d: 184–185.5 °C, yield 86%; lit.5b: 184–185 °C, yield 96%].
13C NMR (100.6 MHz, DMSO-d6): δ = 45.4 (C7a), 49.1 (C3), 51.4 (C7), 81.3 (C6), 87.2 (C3a), 119.3 (C2′ and C6′), 123.8 (C4′), 128.5 (C3′ and C5′), 135.2 and 136.8 (C5 and C4), 139.5 (C1′), 170.2 and 172.6 (C1 and CO2H).
MS (EI, 70 eV): m/z (%) = 271 (19) [M]+, 253 (50), 226 (33), 172 (100), 128 (30), 115 (26), 104 (45), 80 (74), 53 (45), 43 (25).
Anal. Calcd for C15H13NO4: C, 66.41; H, 4.83; N, 5.16. Found: C, 66.21; H, 4.62; N, 5.01.
This compound was obtained and described previously3d,7a,7b [lit.3d: 164 °C, yield 90%; lit.7a: 150 °C, yield 95%; lit.7b: 151–152 °C, yield 76%].
13C NMR (100.6 MHz, DMSO-d6): δ = 44.6 (C7a), 45.3 and 47.6 (C3 and CCH2Ph), 50.2 (C7), 81.1 (C6), 88.4 (C3a), 127.0 (C4′), 127.4 and 128.5 (C2′, C3′, C5′, C6′), 135.7 and 136.6 (C4 and C5), 136.7 (C1′), 170.6 and 172.9 (CO2H and C1).
Anal. Calcd for C16H15NO4: C, C, 67.36; H, 5.30; N, 4.91. Found: C, 67.22; H, 5.11; N, 4.69.
Representative characteristics of isoindoles 3a,3b (all other data for 3c–l are given in the ESI†)
13C NMR (100.6 MHz, DMSO-d6): δ = 51.7 (C1), 121.0 (C2′ and C6′), 125.5, 126.6, 128.6, 131.0, 132.2 (C5, C6, C7, C3′, C4′, C5′), 128.8 and 129.0 (C3a and C4), 137.4 (C1′), 142.2 (C7a), 164.5 and 167.7 (C3 and CO2H).
13C NMR (100.6 MHz, DMSO-d6): δ = 46.4 (CH2Ph), 50.8 (C1), 127.7 (C4′), 128.0 (C2′ and C6′), 128.7 (C3′ and C5′), 128.4 and 128.8 (C3a and C4), 127.7, 131.7, 132.2 (C5, C6, C7), 135.8 (C1′), 143.0 (C7a), 164.7 and 168.9 (C3 and CO2H).
(8aS*,9R*,10S*,12aR*,12bR*)-8-Oxo-5,8,8a,9,10,12b-hexahydro-6H-10,12a-epoxyisoindolo[1,2-a]isoquinoline-9-carboxylic acid (5a) and (8aS*,9R*,10S*,12aR*,12bR*)-2,3-dimethoxy-8-oxo-5,8,8a,9,10,12b-hexahydro-6H-10,12a-epoxyisoindolo[1,2-a]isoquinoline-9-carboxylic acid (5b) were synthesized earlier.11
Method A. A solution of carboxylic acid 5a or 5b (1 g, ∼3 mmol) in orthophosphoric acid (15 mL) was stirred at 80–85 °C for 50 min. After cooling to room temperature, the reaction mixture was poured into water (50 mL). The precipitate formed was filtered off, washed with water (until neutral reaction of rinsing water) and air dried. Further crystallization of crude products from i-PrOH/DMF provided pure compounds 6a,b, as white fine needle-shaped crystals.
Method B. Carboxylic acid 5 (3.6 mmol) was added to a stirred solution of NaOH (1.43 g, 36 mmol) in 15 mL H2O. The reaction mixture was heated under reflux for 1.5 h in argon atmosphere. After cooling to room temperature the reaction mixture was adjusted to pH ∼2–3 with concentrated HCl. The formed precipitate was filtered off, washed with water (3 × 15 mL) and air dried. The crude product was crystallized from i-PrOH/DMF to obtain pure compounds 6a,b.
Method C. A heterogeneous mixture of carboxylic acid 5 (3.6 mmol), and p-TSA (1.9 g, 11 mmol) in toluene (30 mL) was stirred under reflux for 4 h. Then toluene was decanted off and the resulting dark brown, vitriform residue was dissolved in hot CHCl3 (3 × 50 mL) (a considerable part of the substance did not dissolve). The combined organic phases were washed with H2O (3 × 100 mL), then with brine (100 mL), dried over MgSO4, and concentrated in vacuo to give a viscous brown oil, which was purified by column chromatography (EtOAc-CHCl3) to give no identifiable products.
13C NMR (100.6 MHz, DMSO-d6): δ = 28.7 (C5), 35.0 (C6), 86.5 (C12b), 126.6, 127.8, 128.0, 129.1 (C1, C2, C3, C4), 127.2 (C9), 128.8 (C8a), 133.3 and 132.3 (C10 and C11), 128.4 (C12), 136.1 and 134.4 (C12c and C4a), 149.2 (C12a), 164.9 (C8), 166.8 (CO2H).
MS (EI, 70 eV): m/z (%) = 279 (25), 250 (21), 235 (100), 206 (29), 178 (39), 130 (16), 103 (24), 77 (39), 43 (40).
13C NMR (100.6 MHz, DMSO-d6): δ = 28.6 (C5), 35.1 (C6), 55.5 and 56.0 (2 × OMe), 86.3 (C12b), 111.1 and 111.8 (C1 and C4), 126.93, 126.96, 127.7, 128.8 (C4a, C8a, C9, C12c), 127.9, 132.1, 133.3 (C10, C11, C12), 147.6, 149.0, 149.6 (C2, C3, C12a), 164.9 and 166.9 (C8 and CO2H).
MS (EI, 70 eV): m/z (%) = 339 (100), 324 (46), 308 (50), 295 (95), 278 (34), 165 (43), 77 (50), 43 (69).
(3aR*,9aR*,10S*,11R*,13aS*,13bR*,13cR*)-9-Oxo-1,2,9,9a,10,11,13b,13c-octahydro-3aH-11,13a-epoxyfuro[3,2-c]isoindolo[2,1-a]quinoline-10-carboxylic acid (7a) and (4aR*,10aR*,11S*,12R*,14aS*,14bR*,14cR*)-10-oxo-2,3,10,10a,11,12,14b,14c-octahydro-1H,4aH-12,14a-epoxyisoindolo[2,1-a]pyrano[3,2-c]quinoline-11-carboxylic acid (7b) were synthesized earlier.12
Method A. A solution of carboxylic acid 7a or 7b (∼1 g, 2.8 mmol) in 15 mL H3PO4 was stirred at 85 °C for 30 min. After cooling to room temperature, the reaction mixture was poured into water (50 mL). The formed precipitate was filtered off and air-dried. Recrystallization from i-PrOH/DMF produced white needle-shaped crystals of 8 in the form of the single diastereoisomer 8aA, 8bA.
Method B. Carboxylic acid 7 (3.6 mmol) was added to a stirred solution of NaOH (1.44 g, 36 mmol) in H2O (13 mL). The reaction mixture was heated under reflux for 1.5 h. After cooling to room temperature the reaction mixture was adjusted to pH 2–3 with concentrated HCl. The formed precipitate was filtered off, washed with water (2 × 15 mL) and air dried affording a mixture of diastereomers 8aA/8aB, 8bA/8bB. The isomer ratio (according to 1H NMR) is given in Scheme 4. Multiple fractional crystallization of these mixtures from i-PrOH/DMF afforded pure compounds 8aA, 8aB and 8bA, 8bB as individual diastereoisomers.
13C NMR (100.6 MHz, DMSO-d6): δ = 23.9 (C1), 39.1 (C13c), 59.2 (C13b), 65.8 (C2), 74.1 (C3a), 119.1 (C7), 128.1, 129.4, 130.2, 133.7 (C3b, C7a, C9a, C10), 125.2, 125.8, 127.7, 129.7, 131.1, 132.5 (C4, C5, C6, C11, C12, C13), 144.4 (C13a), 164.6 and 166.3 (C9 and CO2H).
13C NMR (100.6 MHz, DMSO-d6): δ = 28.7 (C1), 41.4 (C13c), 58.6 (C13b), 65.6 (C2), 75.6 (C3a), 120.2 (C7), 126.6, 128.8, 130.1, 133.7 (C3b, C7a, C9a, C10), 125.2, 126.5, 128.6, 130.7, 131.0, 132.9 (C4, C5, C6, C11, C12, C13), 145.7 (C13a), 165.63 and 165.60 (C9 and CO2H).
HRMS (DART) m/z (rel. intensity): 322.110 (2) [M+H]+ (100) (exact mass for C19H15NO4 321.1001), 323.12 (27).
Anal. Calcd for C19H15NO4: C, 71.02; H, 4.71; N, 4.36. Found: C, 71.31; H, 4.54; N, 4.57.
13C NMR (100.6 MHz, DMSO-d6): δ = 16.9 (C2), 23.6 (C1), 34.3 (C14c), 59.9 (C3), 61.6 (C14b), 70.1 (C4a), 119.1 (C8), 125.1, 125.6, 127.0, 127.5, 131.1, 132.5 (C5, C6, C7, C12, C13, C14), 125.7, 128.8, 129.4, 134.8 (C4b, C8a, C10a, C11), 143.3 (C14a), 164.6 and 166.1 (C10 and CO2H).
13C NMR (100.6 MHz, DMSO-d6): δ = 21.0 (C2), 23.7 (C1), 38.4 (C14c), 57.1 (C14b), 68.6 (C3), 74.1 (C4a), 119.7 (C8), 125.1, 127.8, 129.0, 130.9, 131.2, 132.6 (C5, C6, C7, C12, C13, C14), 127.3, 129.1, 130.2, 134.5 (C4b, C8a, C10a, C11), 144.4 (C14a), 165.6 and 167.1 (C10 and CO2H).
HRMS (DART) m/z (rel. intensity): 336.126 (3) [M+H]+ (100) (exact mass for C20H17NO4 335.1158), 337.13 (18).
Anal. Calcd for C20H17NO4: C, 71.63; H, 5.11; N, 4.18. Found: C, 71.38; H, 5.50; N, 3.97.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant no. 07-03-00083a).References
- (a) For reviews on isoindole chemistry, see: T. J. Donohoe, In Science of Synthesis; E. J. Thomas Ed.; Georg Thieme Verlag; Stuttgart, 2000, Vol. 10, p. 653 Search PubMed; (b) G. B. Jones and B. J. Chapman, In Comprehensive Heterocyclic Chemistry II, A. R. Katrizky, C. W. Rees, E. F. V. Scriven ed.; Pergamon: Oxford, 1996, Vol. 2, p. 1 Search PubMed; (c) V. A. Kovtunenko and Z. V. Voitenko, Russ. Chem. Rev., 1994, 63, 997–1018 CrossRef; (d) R. Bonnett and S. A. North, Adv. Heterocycl. Chem., 1982, 29, 341 CrossRef; (e) F. S. Babichev, V. A. Kovtunenko and A. K. Tyltin, Russ. Chem. Rev., 1981, 50, 1087–1103 CrossRef; (f) J. D. White and M. E. Mann, Adv. Heterocycl. Chem., 1969, 10, 113 CrossRef CAS.
- (a) Reviews on methods of construction of the [1,2] isoindolo-condensed heterocycles: E. V. Boltukhina, F. I. Zubkov and A. V. Varlamov, Chem. Heterocycl. Compd., 2006, 42, 831–857 CrossRef CAS; (b) E. V. Boltukhina, F. I. Zubkov and A. V. Varlamov, Chem. Heterocycl. Compd., 2006, 42, 971–1001 CrossRef CAS.
- (a) A. V. Varlamov, N. V. Sidorenko, F. I. Zubkov and A. I. Chernyshev, Chem. Heterocycl. Compd., 2002, 38, 490–491 CrossRef CAS; (b) F. I. Zubkov, E. V. Boltukhina, K. F. Turchin and A. V. Varlamov, Tetrahedron, 2004, 60, 8455–8463 CrossRef CAS; (c) V. V. Kouznetsov, F. I. Zubkov, U. M. Cruz, L. G. Voskressensky, L. Y. Vargas Mendez, L. Astudillo and E. E. Stashenko, Lett. Org. Chem., 2004, 1, 37–39 CrossRef CAS; (d) A. V. Varlamov, E. V. Boltukhina, F. I. Zubkov, N. V. Sidorenko, A. I. Chernyshev and D. G. Grudinin, Chem. Heterocycl. Compd., 2004, 40, 22–28 CrossRef CAS; (e) F. I. Zubkov, E. V. Boltukhina, K. F. Turchin, R. S. Borisov and A. V. Varlamov, Tetrahedron, 2005, 61, 4099–4113 CrossRef CAS; (f) V. V. Kouznetsov, U. M. Cruz, F. I. Zubkov and E. V. Nikitina, Synthesis, 2007, 375–384 CrossRef CAS.
- (a) The latest reviews on IMDAF reactions: F. I. Zubkov, E. V. Nikitina and A. V. Varlamov, Russ. Chem. Rev., 2005, 74, 639–669 CrossRef CAS; (b) P. Vogel, J. Cossy, J. Plumet and O. Arjona, Tetrahedron, 1999, 55, 13521–13642 CrossRef CAS; (c) C. O. Kappe, S. S. Murphree and P. Albert, Tetrahedron, 1997, 53, 14179–14233 CrossRef CAS; (d) G. Brieger and J. N. Bennett, Chem. Rev., 1980, 80, 63–97 CrossRef CAS.
- (a) D. Bilović, Ž. Stojanac and V. Hahn, Tetrahedron Lett., 1964, 2071–2074 CrossRef; (b) D. Bilović, Croat. Chem. Acta, 1966, 38, 293–298 ( Chem. Abstr. , 1967 , 66 , 55416 ) Search PubMed; (c) D. Bilović and V. Hahn, Croat. Chem. Acta, 1967, 39, 189–197 ( Chem. Abstr. , 1968 , 68 , 105055 ) Search PubMed; (d) D. Bilović, Croat. Chem. Acta, 1968, 40, 15–22 ( Chem. Abstr. , 1968 , 69 , 486751 ) Search PubMed; (e) A. D. Mance, B. Borovička, B. Karaman and K. Jacopčić, J. Heterocycl. Chem., 1999, 36, 1337–1341 CrossRef CAS; (f) A. D. Mance, B. Borovička, K. Jacopčić, G. Pavlović and I. Leban, J. Heterocycl. Chem., 2002, 39, 277–285 CrossRef CAS.
- (a) J. E. Hernández, S. Fernández and G. Arias, Synth. Commun., 1988, 18, 2055–2061 CrossRef; (b) D. Prajapati, D. R. Borthakur and J. S. Sandhu, J. Chem. Soc., Perkin Trans. 1, 1993, 1, 1197–1200 RSC; (c) L. Sader-Bakaouni, O. Charton, N. Kunesch and F. Tillequin, Tetrahedron, 1998, 54, 1773–1782 CrossRef CAS; (d) R. Murali, H. Surya Prakash Rao and H. W. Scheeren, Tetrahedron, 2001, 57, 3165–3174 CrossRef CAS; (e) A. Padwa, K. R. Crawford, C. S. Straub, S. N. Pieniazek and K. N. Houk, J. Org. Chem., 2006, 71, 5432–5439 CrossRef CAS; (f) X. Huang and J. Xu, J. Org. Chem., 2009, 74, 8859–8861 CrossRef CAS; (g) A. A. Mir, V. V. Mulwad and G. K. Trivedi, J. Heterocyclic Chem., 2010, 47, 214–218 CAS.
- (a) P. S. Sarang, A. A. Yadav, P. S. Patil, U. M. Krishna, G. K. Trivedi and M. M. Salunkhe, Synthesis, 2007, 1091–1095 CAS; (b) C. P. Gordon, N. Byrne and A. McCluskey, Green Chem., 2010, 12, 1000–1006 RSC; (c) D. D. Claeys, C. V. Stevens, B. I. Roman, P. Van De Caveye, M. Waroquier and V. Van Speybroeck, Org. Biomol. Chem., 2010, 8, 3644–3654 RSC.
- (a) A. Pelter and B. Singaram, J. Chem. Soc., Perkin Trans. 1, 1983, 1383–1386 RSC; (b) I. N. Namboothiri, M. Ganesh, S. M. Mobin and M. Cojocaru, J. Org. Chem., 2005, 70, 2235–2243 CrossRef CAS.
- A. Ilyin, V. Kysil, M. Krasavin, I. Kurashvili and A. V. Ivachtchenko, J. Org. Chem., 2006, 71, 9544–9547 CrossRef CAS.
- (a) A. Pelter and B. Singaram, Tetrahedron Lett., 1982, 23, 245–248 CrossRef CAS; (b) G. O. Kachkovskyi and O. I. Kolodiazhnyi, Phosphorus, Sulfur Silicon Relat. Elem., 2009, 184, 890–907 CrossRef CAS.
- F. I. Zubkov, J. D. Ershova, A. A. Orlova, V. P. Zaytsev, E. V. Nikitina, A. S. Peregudov, A. V. Gurbanov, R. S. Borisov, V. N. Khrustalev, A. M. Maharramov and A. V. Varlamov, Tetrahedron, 2009, 65, 3789–3803 CrossRef CAS.
- (a) F. I. Zubkov, V. P. Zaitsev, A. M. Piskareva, M. N. Eliseeva, E. V. Nikitina, N. M. Mikhailova and A. V. Varlamov, Russ. J. Org. Chem., 2010, 46, 1192–1206 CrossRef CAS; (b) F. I. Zubkov, V. P. Zaitsev, A. S. Peregudov, N. M. Mikhailova and A. V. Varlamov, Russ. Chem. Bull., 2007, 56, 1063–1079 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: full physicochemical and spectral data of all synthesized compounds. See DOI: 10.1039/c2ra20295f/ |
| This journal is © The Royal Society of Chemistry 2012 |