Interdependence of structure and chemical order in high symmetry (PdAu)N nanoclusters†
Andrew J.
Logsdail
and
Roy L.
Johnston
*
School of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, U.K. E-mail: r.l.johnston@bham.ac.uk; Fax: +44 (0)121 414 4403; Tel: +44 (0)121 414 7477
First published on 20th April 2012
Abstract
In this work we study the stabilities of high-symmetry AuN, PdN and (AuPd)N clusters, for N < 1500, using mathematical constructs, a semi-empirical potential with two different parameter sets, and a quasi-Newtonian minimisation technique. For PdN clusters, both parameter sets tested result in preferences for icosahedral (Ih) structures for N < 1000 over other high-symmetry 12-vertex geometries; for AuN clusters we find a tendency towards face-centred cubic (FCC) structures at values of N lower than seen for PdN: parameter set I of Cleri and Rosato [Cleri and Rosato, Phys. Rev. B, 1993, 48, 22] gave a transition at N ≈ 650 to the I-Dh, whilst for parameter set II of Baletto et al. [Baletto et al., J. Chem. Phys., 2002, 116, 3856] this value was lower still. For (AuPd)N clusters we found that the preferred arrangement is (PdcoreAushell)N, with thin (monolayer) surface coverings of Au being most energetically favourable compared to the homogeneous clusters; however for parameter set II multiple layers of Au lead to energetic instability. (AucorePdshell)N clusters are not energetically favourable with thin coatings of Pd, however as the shell coating thickens so the stability improves. Ih structures are unfavourable compared to the Ino-decahedron and cuboctahedron for (AucorePdshell)N, whereas the FCC-type structures are strongly preferred for (PdcoreAushell)N. Overall, the strong tendency towards core–shell segregation is emphasised for parameter set I more than II, agreeing with previous work on smaller (AuPd)N clusters.
1 Introduction
Computational modelling plays an important role in understanding the properties of nanoclusters, as it allows the prediction of structures for the lowest energy isomer (i.e. the global minimum, GM),1 as well as providing information on preferential cluster geometries,2 or local minima, and further details such as metal segregation in bimetallic systems.3 A large amount of computational research work has been done previously on both homogeneous and heterogeneous clusters; recent comprehensive reviews have been carried out by Baletto et al.4 and Ferrando et al.3 respectively, and we highlight some of the key publications below.The energetics of small Pd nanoparticles have been thoroughly investigated by several computational groups using first-principle techniques.2,5–10 Besides their catalytic properties, work on these clusters is interesting as smaller Pd clusters have non-zero magnetic moments.11 At large sizes, ab initio calculations are rare due to their computational expense: DFT calculations by Nava et al. found an early preference for bulk-like structures when the number of atoms N > 100, with a larger binding energy (Eb) for FCC geometries than icosahedra (Ih) or Ino-decahedra (I-Dh).5 However at “magic numbers”, Ih and cuboctahedra (CO) remain very energetically competitive, a result confirmed by Guirado-López et al.9 Baletto and Ferrando et al. studied relative geometric stabilities using the Gupta potential12,13 on a wide range of large PdN clusters (N < 5000), showing a trend of Ih for low nuclearities, FCC for large nuclearities and M-Dh in between, with interval windows greater than those observed in a similar study of AuN clusters.2
Small AuN clusters (N ≤ 10) have been thoroughly investigated using ab initio techniques, showing a preference for planar geometries.14–16 Density functional theory (DFT) calculations and IR photodepletion experiments have shown that Au20 is a tetrahedron17,18 with FCC packing; for other sizes low-symmetry structures are known to be energetically competitive.19 For larger clusters ab initio calculations are less feasible: as previously mentioned Baletto and Ferrando carried out calculations on high-symmetry structures using the RGL potential, where a preferential structural interval for Ih was identified for 0 < N < 50, followed by an M-Dh interval of similar size (50 < N < 100).2 After this, crystalline structures were prominent, and similar results were found by various other groups using other semi-empirical potentials.20,21 Li, Palmer, Johnston and co-workers22,23 identified Au309 cluster geometries by comparing experimental scanning transmission electron microscopy (STEM) images with image simulations (based on low energy structures found using the semi-empirical Gupta potential), and found a number of competing structures; Ih, I-Dh and crystalline (FCC-like) geometries were all observed.
Empirical calculations for small (PdAu)N clusters have been conducted by the Johnston group using the Gupta Potential coupled with a genetic algorithm (GA)24 for N < 50,25 and also specifically N = 34 and 38.26 In both these works various potential parameter sets were tested, with parameters derived by Cleri and Rosato12 found to favour PdcoreAushell segregation, whilst more recent parameters derived from ab initio calculations indicate a more mixed configuration; higher level calculations were unable to allow any more definitive opinions on the matter. These findings highlight the sensitivity of structural configurations on the parameter sets used, especially at small sizes.26
Calculations for larger clusters using the embedded-atom model (EAM) showed a preference for PdcoreAushell segregation,27 which has been backed up by classical molecular dynamics (MD) calculations indicating core–shell inversion from AucorePdshell to PdcoreAushell above 500 K,28 the lower surface energy (Esurf) of gold (Au: 96.8 meV Å−2, Pd: 131 meV Å−2![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 29) is thought to be the driving force behind this configuration. Experimental work conducted in the same article reports the successful synthesis of AucorePdshell, PdcoreAushell and mixed PdAu nanoparticles,28 showing that thermodynamic driving forces may be restricted by kinetic trapping, depending on the synthetic method. In larger clusters, Ferrer et al. have reported the experimental formation of tri-layered (onion-like) Au/Pd nanoparticles.30
29) is thought to be the driving force behind this configuration. Experimental work conducted in the same article reports the successful synthesis of AucorePdshell, PdcoreAushell and mixed PdAu nanoparticles,28 showing that thermodynamic driving forces may be restricted by kinetic trapping, depending on the synthetic method. In larger clusters, Ferrer et al. have reported the experimental formation of tri-layered (onion-like) Au/Pd nanoparticles.30
2 Methodology
In this paper, we discuss the relative energetics for different high-symmetry structures composed of Pd, Au, or a combination of the two. Clusters have been created using mathematical constructs, and then energetically minimised; we look to identify stability trends for different compositions and geometries, in order to compare our results with experimental observations.2.1 High symmetry structures
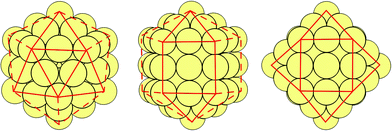
It has been shown that three common high-symmetry 12-vertex geometries, namely Ih, I-Dh and CO, can be arranged from the same “magic number” nuclearities given by the third-order polynomial:31
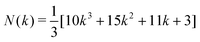
| (1) |
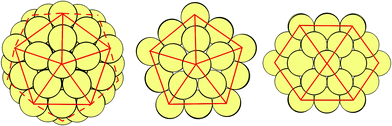
Each of these identified structures (Ih, I-Dh and CO) have distinct atomic arrangements (as shown in Fig. 1 and 2), leading to differing attributes. The Ih consists of 20 (111) faces, making it the most spherical of the three structures. This arrangement minimises the surface area to volume ratio, whilst maximising nearest neighbour contact, at the expense of high strain energy. The Ih geometry is most common for small clusters, where the cost of having large surfaces is too great unless exceptional electronic configurations exist.32 For larger clusters, the minimisation of internal strain energy is more important. The CO is a fragment of an FCC crystal, with 8 (111) faces and 6 (100) faces; strain energy is at its lowest due to the bulk crystal nature, and there are a reasonable number of nearest-neighbour contacts. The CO is most common for large clusters (N > 5000) as we approach the bulk limit. The I-Dh occupies the transitional space between these two geometries, with its structure having neither the tight packing of the Ih structure nor the high surface area of the CO: it has 10 (111) faces and 5 (100) faces, and is most commonly seen for medium sized clusters.
 | ||
| Fig. 1 High symmetry structures with matching magic number nuclearities, illustrated for k = 2 (N = 55). From left to right: Ih, I-Dh and CO. Predominant faces are outlined in solid red lines, whilst background faces are marked with dashed red lines. | ||
 | ||
| Fig. 2 As for Fig. 1, with structures rotated by 90° around an x-axis positioned horizontally across the page. | ||
2.2 The Gupta potential
It remains computationally expensive to perform ab initio calculations on large clusters (N > 100), and so empirical potentials remain widely used. Empirical potentials are derived from the fitting of experimental data to an assumed functional form, with common examples including the Murrell–Mottram,33 Gupta,12 Sutton–Chen34 and EAM.35 When coupled with efficient search techniques (e.g. GAs24) highly effective theoretical tools result.The Gupta potential, derived from Gupta's expression for the cohesive energy (Ecoh) of a bulk material,36 is based on the second moment approximation to tight-binding theory and is constructed from an attractive many-body (Vm) term and a repulsive pair (Vr) term, obtained by summing over all N atoms:
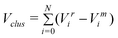 | (2) |
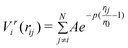 | (3) |
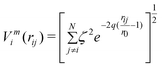 | (4) |
In these equations, rij represents the distance between atoms i and j; and r0 is the nearest neighbour distance in the bulk (in Å). The Gupta potential parameters (A, ζ, p and q) are fitted to the bulk properties of each metal (i.e. Ecoh, bulk modulus, the annihilation of the energy gradient at r0, and in some cases the surface energy, Esurf). After fitting to the bulk properties, one is left with only two independent parameters (p and q), which determine the range of the repulsive and attractive terms, respectively. The parameter sets I and II used in the following work are taken from the work of Cleri and Rosato12 and Baletto et al.,2 respectively, and are shown in Table 1 and 2, and plotted in the ESI.† Parameter sets are not mixed, however arithmetic means are used for bimetallic Au–Pd interactions.
The parameters used by Baletto et al.2 incorporate a long distance cut-off into the potential for atoms further than 2 neighbours’ distance away (>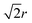 ).37 The interatomic potential [eqn (2)] decays with increasing distance and introducing a cut-off speeds up the energy calculations for large clusters. The implementation of this cutoff is documented in the ESI.† Structure minimisation is achieved using the Limited-Memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm.38
).37 The interatomic potential [eqn (2)] decays with increasing distance and introducing a cut-off speeds up the energy calculations for large clusters. The implementation of this cutoff is documented in the ESI.† Structure minimisation is achieved using the Limited-Memory Broyden–Fletcher–Goldfarb–Shanno (L-BFGS) algorithm.38
3 Energetic analysis
In order to quantify our results, we must introduce several equations that allow us to compare cluster stability. Firstly, we calculate the average binding energy per atom (Eb) using:
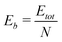
| (5) |
The ratio of excess energy (Eexc) to the number of cluster surface atoms is given as:39,40
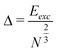
| (6) |
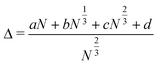 | (7) |
The numerator is still equivalent to Eexc, and in this expression the constant d comes from the strain created by the vertices of the cluster; cN1/3 represents strain from the edges, bN2/3 from surface facets and the volume term aN is due to internal strain. aN vanishes for FCC geometries as N → ∞, as in the bulk crystal structure, whilst strain is present for all other geometries. Minimisation of Δ infers relative overall stability of a given geometry.
4 Results and discussion
High-symmetry structures were systematically created using the mathematical constructs outlined above, before being energetically minimised using the parameters of either Cleri and Rosato12 or Baletto et al.,2 henceforth referred to exclusively as parameter sets I and II, respectively.4.1 (AucorePdshell)N clusters
With the 12-vertex high-symmetry structures it is possible to conduct systematic studies of their core–shell bimetallic structures by building up clusters with layers of different atom types. One of the more fundamental cluster configurations is that of core–shell segregation, whereby one element occupies sites close to the core of the cluster and the other element occupies more surface sites. This kind of arrangement is commonly documented experimentally for (AuPd)N.3,41 By creating a fixed core of one element and nuclearity, and growing shells of the other element on the surface (Fig. 3), we are able to compare the relative stabilities of the core–shell bimetallic systems with respect to the homogeneous monometallic systems, and also the relative stabilities of the different core–shell structures to each other, i.e. the Ih, I-Dh and CO. Naively, one could expect a linear transition between the structural preferences of Au and Pd as we are using arithmetic means for the Au–Pd interaction.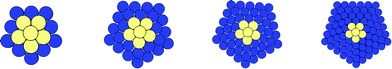 | ||
| Fig. 3 Cross section of (AucorePdshell)N Dh with a fixed core of 13 atoms (k = 1). Left to right: N = 55, 147, 309 and 561, respectively. Pd and Au are represented in blue and yellow, respectively. | ||
Comparison of Δ for the pure clusters offers an initial insight into the predicted energetic evolution with the addition of a second element to the cluster: Fig. 4 shows plots of Δ for the Ih, I-Dh and CO values for AuN and PdN clusters. For parameter set I we notice that for N < 150 the Aun structures have lower Δ values than PdN; however, this does not hold true for N > 200, with PdN becoming energetically preferred and the gap between the trend lines increasing up to N = 1500. For parameter set II the AuN structures remain more energetically stable than PdN, with the gap between the two remaining constant at ∼0.5 eV. The Ih structure is favoured up to large N for PdN, unlike AuN, and this is shown for both parameter sets: these results have been seen previously2 thus a more detailed analysis is offered in the ESI.†
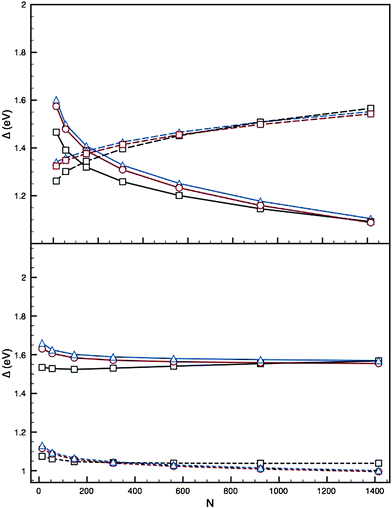 | ||
| Fig. 4 Top: Plot of Δ against N for the 12-vertex high-symmetry structures, using parameter set I of Cleri and Rosato.12 Bottom: as above, using parameter set II of Baletto et al.2 Ih (black squares), I-Dh (red circles) and CO (blue triangles) are shown, with PdN (solid lines) and AuN (dashed lines) plotted. | ||
Ih, I-Dh and CO clusters were generated with fixed-size Au cores and Pd layers added to form the shell, as illustrated in Fig. 3. Results from parameter sets I and II have been plotted in Fig. 5 and 6, respectively. In Fig. 5, comparison with PdN shows that adding the Au impurity results in an increase in Eexc, and thus Δ, for all clusters; and the greater the concentration of Au in the core of the structure, the greater the difference is between Δ for PdN and (AucorePdshell)N. Though not plotted, comparison with AuN also shows that Δ for (AucorePdshell)N is increased with respect to AuN when the number of Pd layers is small (k < 4), making this form of mixing unfavourable for bimetallic systems at low nuclearities. Larger clusters appear more stable, with Δ for the bimetallic cluster falling between the pure clusters, and the difference in Δ between PdN and (AucorePdshell)N decreasing as N increases. We also note that for a core of Au55 or greater, Ih is no longer the preferred structure with respect to the I-Dh and CO; this is in agreement with the observed trends for the pure AuN clusters.
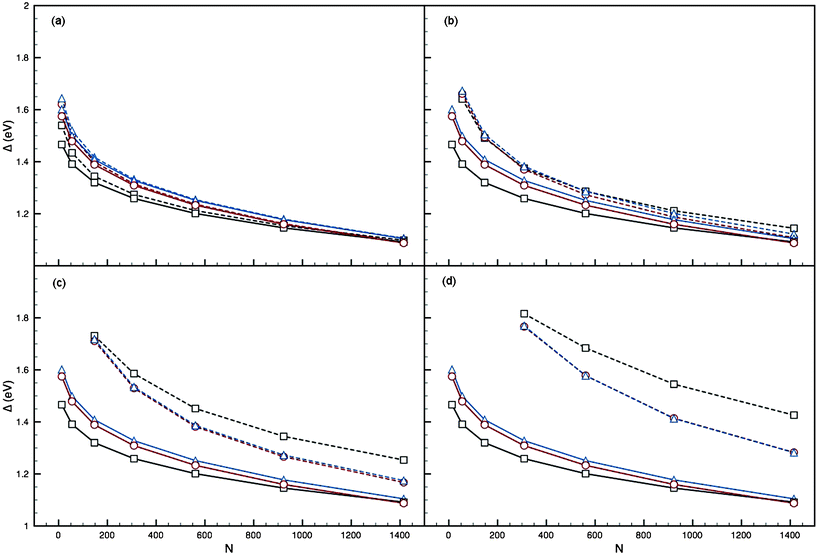 | ||
| Fig. 5 Plots of Δ against N for the AucorePdshell 12-vertex high-symmetry structures (dashed lines), using parameter set I of Cleri and Rosato,12 with cores: (a) Au1 (b) Au13 (c) Au55 (d) Au147. Ih (black squares), I-Dh (red circles) and CO (blue triangles) are shown, with PdN (solid lines) also plotted. | ||
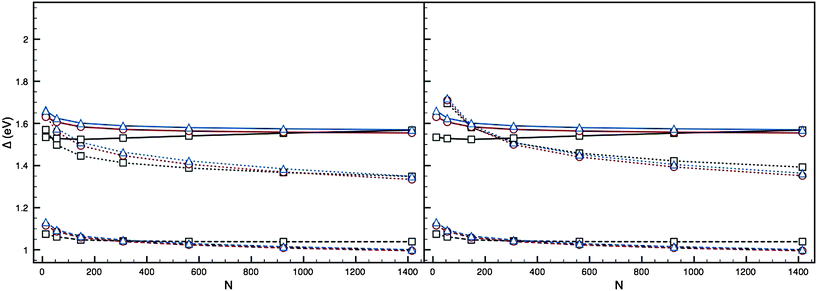 | ||
| Fig. 6 Plots of Δ against N for the AucorePdshell 12-vertex high-symmetry structures (dotted lines), using parameter set II of Baletto et al.,2 with cores: (a) Au1 and (b) Au13. Ih (black squares), I-Dh (red circles) and CO (blue triangles) are shown, with PdN (solid lines) and AuN (dashed lines) also plotted. | ||
For parameter set II (Fig. 6), this destabilisation is not so prominent. Whilst thin (monolayer) Pd shells are not energetically favourable, especially with increasing size of the AuN core, multiple Pd layers have a quenching effect on this disparity, reducing Eexc such that for an Au13 core with 3 layers of Pd or greater (k > 3) it appears more energetically favourable than pure PdN clusters.
4.2 (PdcoreAushell)N clusters
Using parameter set I, and inverting the core–shell segregation to form (PdcoreAushell)N, leads to the reverse effect to that witnessed for the (AucorePdshell)N: the value of Δ tends to decrease with respect to the pure clusters (figure provided in the ESI†) as the size of the Pd core increases. The effect is greatest for smaller clusters, with monolayers of Au, with the difference between Δ for the core–shell and for the pure clusters reducing as N increases, for a fixed core. For multiple Au shell layers, Δ shifts to a value between the pure values, illustrating again the averaging effect mentioned previously. We also see that the Ih structure becomes distinctly more stable than the I-Dh and CO structures, whilst the energy gap between the I-Dh and CO also increases. For a core of Pd55 or greater, the transition from Ih to I-Dh and CO is out of the nuclearity range studied (i.e. N > 1500).For parameter set II, the effect of multiple Au layers is not so favourable (Fig. 7). Small Pd impurities in the core of small Au clusters are energetically favourable compared to the pure PdN clusters, but not compared to the AuN clusters. For all core sizes studied, an Au coating becomes unfavourable with respect to the pure clusters when thicker than a monolayer, and increases with thicker Au coverage, illustrating that the segregation we have used may be less stable than the mixed alloy for these parameters, similarly to results seen by Ismail and Johnston.26 The Ih, I-Dh and CO structures are all competitive throughout.
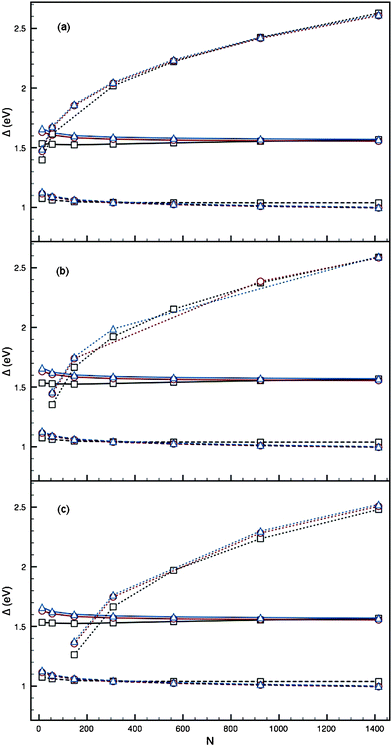 | ||
| Fig. 7 Plots of Δ against N for the PdcoreAushell 12-vertex high-symmetry structures (dotted lines), using parameter set I of Baletto et al.,2 with cores: (a) Pd1 (b) Pd13 and (c) Pd55. Ih (black squares), I-Dh (red circles) and CO (blue triangles) are shown, with AuN (dashed lines) and PdN (solid lines) also plotted. | ||
Attention to the relative stability of the (PdcoreAushell)N structures with just a monolayer coating, as shown in Fig. 9, reveals an unusually high stability, which increases with N (Fig. 8) for both parameter sets I and II.
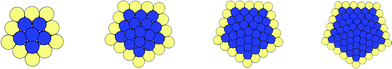 | ||
| Fig. 8 Cross section of (PdcoreAushell)N Dh with a monolayer covering on the shell. Left to right: N = 55, 147, 309 and 561, respectively. Pd and Au are represented in blue and gold, respectively. | ||
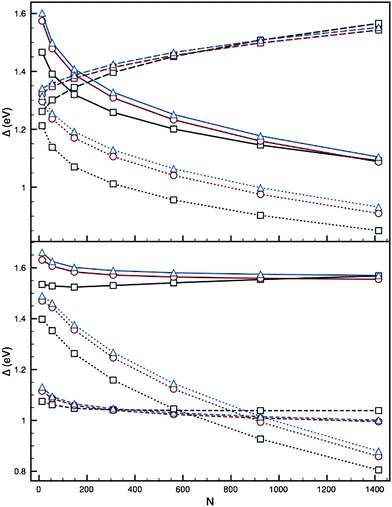 | ||
| Fig. 9 Top: Plots of Δ against N for the AucorePdshell 12-vertex high-symmetry structures, using parameter set I of Cleri and Rosato,12 with a monolayer covering of Au. Bottom: as above, using parameter set II of Baletto et al.41 Ih (black squares), I-Dh (red circles) and CO (blue triangles) are shown; PdN (solid lines), AuN (dashed lines) and PdcoreAushell (dotted lines) are plotted. | ||
A monolayer coating of Au on a Pd core, with parameter set I, results in a value of Δ lower than that for both the AuN and PdN clusters of the same nuclearity. This is a strong indication of the greater stability of the PdcoreAushell segregation, and agrees with the common discovery of core–shell structures in previous structural searches.25,26 The difference between the energies of PdN and (PdcoreAushell)N remains constant as k increases, at a gap of ∼0.3 eV. The Ih structure is energetically preferred throughout, in stark contrast to the same calculations for the AuN clusters, which show an early tendency to adopt crystalline structures.
For parameter set II, a monolayer coating has a similar effect in stabilising the PdN cluster. However, for N < 800 (k < 6) the (PdcoreAushell)N clusters are less stable than the pure AuN clusters. For N > 800, the core–shell segregation becomes more energetically favourable than the pure AuN cluster; this implies that this segregation is disfavoured at low N, where mixing is perhaps preferred, but as N increases, and so the ratio of Pd to Au atoms increases, so does the relative stability of core–shell segregation to this structure. This is perhaps the driving force behind the structural rearrangements seen in previous molecular dynamics studies,28 where core–shell inversion occurs. As the gradient of Δ is greater for the (PdcoreAushell)N cluster than for the pure clusters, we can predict that for N > 1500 the core–shell structural arrangement may become even more favourable, as it minimises Eexc.
5 Conclusions
We have studied the stabilities of high-symmetry AuN, PdN and (AuPd)N clusters, using mathematical constructs, a semi-empirical potential with two different parameter sets, and a quasi-Newtonian minimisation technique. The structures for which calculations were performed include the Ih, I-Dh, and CO; this list is by no means exhaustive but it does give a good insight into the preferred nanocluster packing in high-symmetry structures.For PdN clusters, both parameter sets tested result in preferences for the Ih structures for N < 1000 over other high-symmetry 12-vertex geometries. For AuN clusters, we found a tendency towards FCC structures at values of N lower than those seen for PdN: the parameters of Cleri and Rosato gave a transition at N ≈ 650 from Ih to I-Dh, whilst for the parameters of Baletto et al. this value was lower still.
For (AuPd)N clusters we found that the preferred arrangement is for (PdcoreAushell)N for both parameter sets, with thin (monolayer) surface coverings of Au being the most energetically favoured compared to the homogeneous clusters, and Ih structures proving the most stable throughout; the stability of the Ih structure for the (PdcoreAushell)N cluster at large N is surprising as it is not preferred for the pure clusters above N ≈ 650. The fact that Ih is stabilised at larger sizes for PdcoreAushell than for pure Pd or Au clusters may be because the Pd nearest-neighbour distances are slightly smaller than Au; bulk nearest-neighbour distances are 2.75 Å and 2.88 Å for Pd and Au, respectively (see Table 1 and 2). This could relieve some of the compressive strain and extend the stability range of the Ih structure, as has been seen previously for Cu-doped Au icosahedra.45 The core–shell segregation observed is highly compatible with the experimental measurements Esurf (Pd) > Esurf (Au) and Ecoh (Pd) > Ecoh (Au). For the parameters of Baletto et al. multiple layers of Au lead to energetic instability, with Δ values greater than those for either of the pure metal nanoparticles; this is not the case for the parameters of Cleri and Rosato. (AucorePdshell)N clusters are not energetically favourable with thin coatings of Pd, however as the shell coating thickens so the stability improves. This effect is more prominent for the parameters of Cleri and Rosato. Ih structures are not favourable compared to the I-Dh and CO for (AucorePdshell)N, whereas they are strongly preferred for (PdcoreAushell)N: this trend strengthens as the size of the core increases. Overall, the strong tendency towards core–shell segregation is emphasised for the parameters of Cleri and Rosato,12 and not so much for the parameters of Baletto et al.,2 agreeing with previous work on smaller (AuPd)N clusters.25,26
Exploring other high-symmetry homotops42 for the bimetallic (AuPd)N clusters is necessary in the future, to determine whether other segregated formations are more stable than those described here, with investigation into other lower-symmetry structures also required to give a more complete treatment of structural preference in any future work. From these calculations we will take stable structures forward for electronic analysis using higher-level calculations, as chemical ordering is shown to have an effect on catalytic properties.43
Acknowledgements
AJL acknowledges financial support from the EPSRC, UK (DTA Award Reference: EP/P504678/1) and the University of Birmingham, UK. RLJ acknowledges financial support from the EPSRC, UK. The authors acknowledge support from COST Action MP0903 “Nanoalloys as Advanced Materials: From Structure to Properties and Applications”. The authors acknowledge Prof. Riccardo Ferrando for helpful conversations and supplying some cluster coordinates. The computations described in this article were performed using the University of Birmingham's BlueBEAR HPC service, which was purchased through HEFCE SRIF-3 funds.44References
- R. L. Johnston, Atomic and Molecular Clusters, Taylor and Francis, London, 2002 Search PubMed.
- F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti and C. Mottet, J. Chem. Phys., 2002, 116, 3856–3863 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371–423 CrossRef CAS.
- P. Nava, M. Sierka and R. Ahlrichs, Phys. Chem. Chem. Phys., 2003, 5, 3372–3381 RSC.
- G. Valerio and H. Toulhoat, J. Phys. Chem. A, 1997, 101, 1969–1974 CrossRef CAS.
- B. Kalita and R. C. Deka, J. Chem. Phys., 2007, 127, 244306–244316 CrossRef.
- D. R. Jennison, P. A. Schultz and M. P. Sears, J. Chem. Phys., 1997, 106, 1856–1862 CrossRef CAS.
- R. Guirado-López, M. C. Desjonquères and D. Spanjaard, Phys. Rev. B, 2000, 62, 13188–13195 CrossRef.
- F. Aguilera-Granja, A. Vega, J. Rogan and G. Garcia, Nanotechnology, 2007, 18, 365706–365711 CrossRef.
- N. Watari and S. Ohnishi, Phys. Rev. B, 1998, 58, 1665–1677 CrossRef CAS.
- F. Cleri and V. Rosato, Phys. Rev. B, 1993, 48, 22–33 CrossRef CAS.
- V. Rosato, M. Guillope and B. Legrand, Philos. Mag. A, 1989, 59, 321–336 CrossRef.
- G. Bravo-Perez, I. L. Garzan and O. Novaro, Chem. Phys. Lett., 1999, 313, 655–664 CrossRef CAS.
- J. Wang, G. Wang and J. Zhao, Phys. Rev. B, 2002, 66, 35418–35424 CrossRef.
- V. Bonačić-Koutecký, J. Burda, R. Mitrić, M. Ge, G. Zampella and P. Fantucci, J. Chem. Phys., 2002, 117, 3120–3131 CrossRef.
- J. Li, X. Li, H. J. Zhai and L. S. Wang, Science, 2003, 299, 864–867 CrossRef CAS.
- J. Wang, G. Wang and J. Zhao, Chem. Phys. Lett., 2003, 380, 716–720 CrossRef CAS.
- K. Michaelian, N. Rendón and I. L. Garzón, Phys. Rev. B, 1999, 60, 2000–2010 CrossRef CAS.
- C. L. Cleveland, U. Landman, M. N. Shafigullin, P.W. Stephens and R. L. Whetten, Z. Phys. D, 1997, 40, 503–508 CrossRef CAS.
- N. T. Wilson and R. L. Johnston, Eur. Phys. J. D, 2000, 12, 161–169 CrossRef CAS.
- Z. Y. Li, N. P. Young, M. D. Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46–48 CrossRef CAS.
- B. C. Curley, R. L. Johnston, N. P. Young, Z. Y. Li, M. D. Vece, R. E. Palmer and A. L. Bleloch, J. Phys. Chem. C, 2007, 111, 17846–17851 CAS.
- R. L. Johnston, Dalton Trans., 2003, 4193 RSC.
- F. Pittaway, L. O. Paz-Borbón, R. L. Johnston, H. Arslan, R. Ferrando, C. Mottet, G. Barcaro and A. Fortunelli, J. Phys. Chem. C, 2009, 113, 9141–9152 CAS.
- R. Ismail and R. L. Johnston, Phys. Chem. Chem. Phys., 2010, 12, 8607–8619 RSC.
- B. Shan, L. Wang, S. Yang, J. Hyun, N. Kapur, Y. Zhao, J. B. Nicholas and K. Cho, Phys. Rev. B, 2009, 80, 035404 CrossRef.
- H. B. Liu, U. Pal, A. Medina, C. Maldonado and J. A. Ascencio, Phys. Rev. B, 2005, 71, 075403 CrossRef.
- A. R. Miedema, Z. Metal., 1978, 69, 287–292 CAS.
- D. Ferrer, A. Torres-Castro, X. Gao, S. Sepúlveda-Guzmán, U. Ortiz- Méndez and M. José-Yacamán, Nano Lett., 2007, 7, 1701 CrossRef CAS.
- T. P. Martin, Phys. Rep., 1996, 273, 199–241 CrossRef CAS.
- X. Xing, B. Yoon, U. Landman and J. H. Parks, Phys. Rev. B, 2006, 74, 165423 CrossRef.
- J. N. Murrell and R. E. Mottram, Mol. Phys., 1990, 69, 571–585 CrossRef CAS.
- A. P. Sutton and J. Chen, Philos. Mag. Lett., 1990, 61, 139–146 CrossRef.
- M. S. Daw and M. I. Baskes, Phys. Rev. B, 1984, 29, 6443–6453 CrossRef CAS.
- R. P. Gupta, Phys. Rev. B, 1981, 23, 6265–6270 CrossRef CAS.
- R. Ferrando, Personal Communication, 2009 Search PubMed.
- C. Kittel, Introduction To Solid State Physics, Wiley, New York, 6th edn, 1986 Search PubMed.
- C. L. Cleveland and U. Landman, J. Chem. Phys., 1991, 94, 7376–7396 CrossRef CAS.
- J. Uppenbrink and D. J. Wales, J. Chem. Phys., 1992, 96, 8520–8534 CrossRef CAS.
- R. L. Johnston and R. Ferrando, Faraday Discuss. , 2008, 138, 1–442 RSC.
- J. Jellinek and E. B. Krissinel, Chem. Phys. Lett., 1996, 258, 283–292 CrossRef CAS.
- H. Zhang, T. Watanabe, M. Okumura, M. Haruta and N. Toshima, Nature Mater., 2011, 11, 49–52 CrossRef.
- http://www.bear.bham.ac.uk/bluebear/. , accessed 2012.
- N. T. Wilson and R. L. Johnston, J. Mater. Chem., 2002, 12, 2913–2922 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20309j/ |
| This journal is © The Royal Society of Chemistry 2012 |