Supercritical fluid technology as a new strategy for the development of semi-covalent molecularly imprinted materials
Mara
Soares da Silva
,
Raquel
Viveiros
,
Ana
Aguiar-Ricardo
,
Vasco D. B.
Bonifácio
and
Teresa
Casimiro
*
REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, FCT, Universidade Nova de Lisboa, 2829-516, Caparica, Portugal. E-mail: teresa.casimiro@fct.unl.pt
First published on 9th March 2012
Abstract
Molecularly imprinted polymeric particles with molecular recognition towards Bisphenol A (BPA) were synthesized for the first time using the semi-covalent imprinting approach in supercritical carbon dioxide (scCO2). The material's affinity to BPA was achieved by co-polymerizing ethylene glycol dimethacrylate (EGDMA) with a template-containing monomer, Bisphenol A dimethacrylate (BPADM) in scCO2. Bisphenol A is then cleaved from the polymeric matrix by hydrolysis with tetrabutylammonium hydroxide (n-Bu4OH) also in a supercritical environment, taking advantage of the high diffusivity of scCO2. The selectivity of the molecular imprinted polymer (MIP) was assessed by evaluating its capability to bind BPA in comparison with progesterone and α-ethinylestradiol. In addition, the cross-linked particles were used to prepare a PMMA-based hybrid imprinted membrane by a scCO2-assisted phase inversion method. Results show that the incorporation of MIP particles was able to confer molecular affinity to BPA to the membrane and that at dynamic conditions of filtration, this imprinted porous structure was able to adsorb a higher amount of BPA than the corresponding non-imprinted hybrid membrane. Our work represents a valuable greener alternative to conventional methods, for the synthesis of affinity materials which are able to maintain molecular recognition properties in water.
Introduction
Molecularly imprinted polymers (MIPs) are synthetic matrices with enhanced affinity to certain target molecules. Molecular imprinting uses the functionality of the target molecule (template), to assemble its own recognition site by forming specific interactions with the matrix during polymerization.1 These affinity polymers have association constants comparable to natural receptors but are capable of withstanding harsher conditions of temperature, extreme pH2 and pressure.3 MIPs are also less expensive to synthesize and can be manufactured in large quantities with good reproducibility. These properties have led to numerous reported applications in diverse areas such as in separation processes, synthesis and catalysis, chromatography, sensing, etc.4Conventional methods to synthesize MIPs, typically yield monolithic polymers which have to be crushed, ground and sieved, leading to product loss and resulting in irregular particles in both shape and size with binding sites partly destroyed.5 Recent advances in molecular imprinting techniques include the development of new preparation methods, such as Pickering emulsions,6in situ multi-step swelling and suspension polymerization.7
However, they still show limitations, such as complicated procedures, excessive use of organic solvents and time-consuming purification and drying steps. In addition, it is difficult to prepare MIPs in water because the high concentration of water molecules destroys the polar interactions between the functional monomer and the template, thus organic solvents are typically used.8
The increasing restrictions in the use of organic solvents and the need to overcome the disadvantages of conventional methods, such as the need for crushing and sieving, whilst enhancing the template-desorption from the imprinted matrix, have prompted us to explore the use of supercritical fluid technology in the development of MIPs.
Supercritical CO2 is a suitable porogen for molecular imprinting since it is an apolar aprotic porogen which can stabilize the template-monomer complexes giving rise to materials with high affinity. Furthermore, the high diffusivity of scCO2 provides an ideal medium to extract the template from the formed cavities at the end of the synthesis. In addition, MIPs synthesized using supercritical fluid technology are obtained as solvent free-flowing powders with controlled morphology and porosity.13
In previous studies we have demonstrated that supercritical CO2-assisted non-covalent molecular imprinting is a clean and one-step synthetic route for the preparation of affinity polymeric materials, with attested performance in chromatography,9 drug delivery10–12 and adsorption.13 Our work represents a valuable alternative for the synthesis of materials which can maintain molecular recognition in a water environment.
Herein, we provide further information on the consolidation of supercritical fluid technology in the development of molecular imprinted materials and report, for the first time, the development of a semi-covalent MIP with water-compatible molecular recognition performance, completely processed in a supercritical environment.
Three approaches are typically used to prepare MIPs, covalent, non-covalent and semi-covalent, which differ in the nature of the interactions formed between the template and the functional groups.14 By means of the semi-covalent approach a single molecule is used instead of using two different molecules, such as template and monomer. The template possesses a polymerizable counterpart that reacts with the cross-linker agent yielding the affinity polymer. At the end of the polymerization the template is removed from the matrix by cleavage and the binding sites become available to future rebinding through hydrogen bonds. The semi-covalent approach combines both the strict control of functional group location and uniform distribution, characteristic of covalent imprinting, and the reduced kinetic restriction during rebinding, characteristic of non-covalent imprinting. Due to the coupled advantages, semi-covalently imprinted polymers usually show efficient rebinding.
BPA is an endocrine disruptor that is intensively used in the production of polycarbonate plastics and epoxy resins, with a worldwide production of approximately 2.2 million tonnes in 2009 alone.15 It is known that exposure to small daily doses of BPA increases the risk of breast and prostate cancer16 and diabetes.17 Within the last decades, many efforts have been made to drastically reduce the levels of BPA in environmental waters and soil, through the use of more efficient processes of adsorption, solvent extraction, membrane separation technology and photo degradation. The use of MIPs in the treatment of aqueous solutions provides the affinity adsorption of traces of organic compounds from the samples, selectively removing the target molecules.
In this work, a semi-covalent MIP with enhanced affinity to Bisphenol A (BPA) was prepared by co-polymerizing Bisphenol A dimethacrylate (BPADM) and ethylene glycol dimethacrylate (EGDMA) in scCO2. After template cleavage with n-Bu4NOH in scCO2, the adsorption selectivity of the matrices to BPA, progesterone (PRO) and α-ethinylestradiol (EE), was attested in an aqueous environment. Further immobilization of the cross-linked particles to prepare a hybrid imprinted membrane using a scCO2-assisted phase inversion method was carried out by blending the pre-synthesized polymers within a PMMA casting solution.
Experimental section
Materials
Bisphenol A (BPA, 99% purity) as the analyte of interest, Bisphenol dimethacrylate (BPADM, 99% purity) as template-monomer, methacrylic acid (MAA, 99% purity) as functional monomer and ethylene glycol dimethacrylate (EGDMA, 98% purity) as cross-linker were purchased from Sigma-Aldrich. Azobis(isobutyronitrile) (AIBN, 98% purity) from Fluka was used as initiator. Tetrabutylammonium hydroxide (n-Bu4NOH) 1.0 M solution in MeOH, as the cleavage agent and poly(methyl methacrylate) (PMMA) (molecular weight 996![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were obtained from Sigma-Aldrich. Progesterone (PRO, 99% purity) and α-ethinylestradiol (EE, 98% purity) were purchased from Sigma-Aldrich. Dimethylformamide (DMF, 99.8% purity) was purchased from Riedel-de Haën and acetonitrile and methanol isocratic HPLC grade (99.7% purity) were obtained from Scharlau. Carbon dioxide was obtained from Air Liquide with purity better than 99.998%. All chemicals were used as received without further purification.
000) were obtained from Sigma-Aldrich. Progesterone (PRO, 99% purity) and α-ethinylestradiol (EE, 98% purity) were purchased from Sigma-Aldrich. Dimethylformamide (DMF, 99.8% purity) was purchased from Riedel-de Haën and acetonitrile and methanol isocratic HPLC grade (99.7% purity) were obtained from Scharlau. Carbon dioxide was obtained from Air Liquide with purity better than 99.998%. All chemicals were used as received without further purification.
scCO2-assisted molecular imprinting polymerization
Polymerization reactions in scCO2 were carried out as described elsewhere.18 In a typical reaction using acetonitrile as co-solvent, 3 mL of the organic modifier (10 wt% with respect to CO2) were loaded into the high-pressure cell. To synthesize the semi-covalent MIP, 1.26 mmol of BPADM, 12.58 mmol of EGDMA and 1 wt% of the radical initiator AIBN were introduced into a 33 mL stainless-steel high-pressure cell equipped with two aligned sapphire windows and a Teflon coated magnetic stirrer bar inside. The cell was immersed in a thermostated water bath at 65 °C, CO2 was added up to 21 MPa and the reaction proceeded for 24 hours with constant stirring. At the end of the reaction, the polymer was slowly washed with fresh high-pressure CO2 for 1 hour in order to remove any unreacted residues. As control, the non-imprinted polymer (NIP) composed of methacrylic acid (MAA) and EGDMA was synthesized and processed using the same experimental conditions.High pressure-assisted template removal
Common procedures for BPA cleavage from imprinted polymers use conventional acidic or basic conditions.19,20 Herein, we report for the first time, the hydrolysis of the BPA ester from a semi-covalent MIP cross-linked matrix in supercritical CO2, taking advantage of the high diffusivity and low viscosity of the medium to enhance mass transfer. The successful hydrolysis of polypeptide esters by tetraalkylammonium hydroxide strong bases is reported in the literature.21 This led us to explore the use of a 1 M methanolic solution of tetrabutylammonium hydroxide to accomplish the hydrolysis of P(BPADM-co-EGDMA). The use of an n-Bu4NOH–methanol–scCO2 system as an alternative hydrolysis for highly cross-linked BPA-imprinted polymers comprises three crucial features in the hydrolysis step: (i) n-Bu4NOH acts as the source of hydroxide, (ii) the diffusivity power of CO2 increases the efficiency of the hydrolysis of a highly cross-linked matrix, and (iii) the high solvent power of methanol for BPA enhances its desorption from the matrix. In a typical experiment to cleave BPA from the polymer, P(BPADM-co-EGDMA) (0.925 g), n-Bu4NOH 1.0 M in methanol (1.6 mmol), and a magnetic stir bar were introduced into a high-pressure cell. The cell was immersed in a thermostated water bath at 65 °C and pressurized with CO2 until a final pressure of 20 MPa was reached. Fig. 1 illustrates the cleavage step scheme. After 24 h of reaction the polymer was washed with fresh CO2 (20 MPa) for 1 h. Methanol was added to the polymer and the suspension was filtered under vacuum. Quantification of BPA present in methanol indicated an extraction yield of ca. 39%.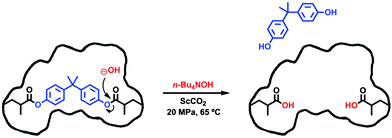 | ||
| Fig. 1 Scheme of the BPA cleavage mechanism in scCO2. | ||
Semi-covalent hybrid membrane preparation
Hybrid imprinted membranes were prepared using a scCO2-assisted phase inversion method.13 Briefly, a hybrid casting solution with 30 wt% of polymer blend consisting of 70:30 of PMMA and MIP or NIP, in 5 mL of dimethylformamide was loaded into a Teflon cap and placed inside the high-pressure cell. The membrane was prepared at 45 °C by immersing the cell in a thermostated water bath, heated by means of a controller (Hart Scientific, Model 2200) that maintained the temperature within ±0.01 °C. Carbon dioxide was added, using a Gilson 305 piston pump, until an operational pressure of 20 MPa was reached. Pressure was set at 20 MPa by means of a back pressure regulator (Jasco BP-2080 plus), which separated the CO2 from the dimethylformamide present in the casting solution. The pressure inside the system was monitored with a pressure transducer (Setra Systems Inc, Model 204) with a precision of ±0.100 kPa. All the experiments were performed with a CO2 flow of 9.8 g min−1 for 3 hours. At the end, the system was slowly depressurized over 20 min and a thin homogeneous membrane was obtained.
Morphological, physical and mechanical characterization of the synthesized materials
The morphology of the synthesized copolymers and corresponding hybrid membranes was characterized using scanning electron microscopy (SEM) in a Hitachi S-2400 instrument, with an accelerating voltage set to 15 kV. For cross-section analysis the membrane samples were frozen and fractured in liquid nitrogen. Samples were mounted on aluminium stubs using carbon tape and were gold/platinum coated. Specific surface area and pore diameter of the polymeric particles were determined by N2 adsorption according to the BET method. An accelerated surface area and porosimetry system (ASAP 2010 Micromeritics) was used under nitrogen flow. The contact angle of the membranes was measured with Millipore water droplets in a KSV Goniometer model CAM 100 at room temperature. The water flux of the membranes was determined using a 10 mL filtration unit (Amicon Corp., model 8010) with an effective area of 4.1 cm2. All the experiments were carried out whilst varying the applied hydrostatic pressure from 0 to 5 bar. The tensile properties of the hybrid membranes were tested by dynamic mechanical analysis (DMA) with a tensile testing machine (MINIMAT firm-ware v.3.1) at room temperature. The samples were cut into 5 mm × 15 mm strips. The length between the clamps was set at 5 mm and the speed of testing was set to 0.1 mm min−1. A full scale load of 20 N and maximum extension of 35 mm were used. Measurements were performed with dried membranes. Load extension graphs were obtained during testing and converted to stress–strain curves. The Young's modulus determined and other characteristic parameters concerned with the properties of the materials are listed in Table 1.| Analysis | Material | |||
|---|---|---|---|---|
| NIP | MIP | PMMA NIP | PMMA MIP | |
| Nitrogen porosimetry | ||||
| BET surface area (m2 g−1) | 58.2 | 49.5 | — | — |
| Pore volume (cm3 g−1) | 0.07 | 0.06 | — | — |
| Average pore diameter (nm) | 5.0 | 4.6 | — | — |
| Contact angle (°) | — | — | 81.6 ± 4.1 | 97.1 ±2.7 |
| Young's modulus (MPa) | — | — | 0.34 ± 0.06 | 0.55 ± 0.09 |
| Water flux (L m−2 h−1 bar−1) | — | — | 25.8 ± 0.8 | 8.8 ± 2.1 |
Analyte adsorption quantification
Batch binding experiments were carried out to evaluate the ability of the synthesized polymers to adsorb BPA, PRO or EE, from aqueous solutions. The polymers (20 mg) were added to 50 mL of aqueous solutions of BPA (5–50 μM) and stirred at 50 rpm for 24 hours. For the binding tests of PRO and EE, given their low water solubility, 0.2% (v/v) of acetonitrile was added to the aqueous solution and concentrations in the range of 5–16 μM were tested. As the concentrations were lower, in order to compare the results of PRO and EE with the results of BPA, lower amounts of polymer were used. By keeping the ratios between the analyte in solution and the weight of polymer constant, the results are comparable and independent of the concentration. Equilibrium was achieved in 24 hours, as confirmed by the quantification of free analyte in solution. The amount of substrate adsorbed by the matrices was assessed through eqn (1), where [S] corresponds to the amount of analyte bound, C0 represents the initial molar concentrations of the analyte, Ct corresponds to the concentrations at predetermined time intervals, V represents the volume of the solution and W corresponds to the weight of the polymeric sample. The samples collected were quantified by UV spectroscopy at 275, 248 and 278 nm for BPA, PRO and EE, respectively. All the experiments were carried out in duplicate.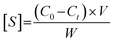 | (1) |
Scatchard analysis
Data from the equilibrium BPA adsorption experiments were processed using the Scatchard equation, where Ka is the association constant, Bmax the apparent maximum binding capacity, Ce represents the free concentration of substrate in equilibrium and B corresponds to the amount of BPA bound to the polymer.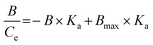 | (2) |
Analyte filtration experiments
To assess the performance of the hybrid membranes to adsorb BPA, PRO and EE in dynamic conditions, PMMA MIP and PMMA NIP were placed separately in the permeability apparatus and hydrostatic pressure was adjusted to assure a constant filtration flow rate of 0.33 mL min−1. Prior to the adsorption experiments, the membranes were equilibrated with 30 mL (3 × 10 mL) of distilled water. The filtration unit was then loaded with 30 mL of an aqueous solution containing 10 μM of BPA and the amount of template adsorbed in the membrane was quantified. The selectivity of the membranes in dynamic conditions was assessed by loading the filtration unit with 10 μM of PRO and EE. The membranes were easily restored by washing with 10 mL of methanol at the same flow rate and no loss of binding capacity was observed. All samples collected were quantified by UV spectroscopy. The amount of analyte bound to the membranes was determined using eqn (1). All the experiments were carried out in triplicate.Results and discussion
Imprinted and non-imprinted polymers were obtained as dry, free-flowing powders in high yields (∼90%, determined gravimetrically) and their morphology was assessed by scanning electron microscopy. Fig. 2 shows the SEM images of MIP and hybrid PMMA MIP. Both MIP and NIP appear as aggregates of smooth surface discrete nanoparticles. The successful hybridization of the membranes can be visualized, with the cross-linked particles homogeneously distributed within the matrix. Table 1 shows the physical and mechanical properties of the copolymers and hybrid membranes prepared. The copolymers have similar physical properties, although MIP has a slightly lower surface area. With regard to the membranes, PMMA MIP presents a higher hydrophobicity than the corresponding PMMA NIP, reflected by a higher contact angle and lower water flux. | ||
| Fig. 2 Scanning electron microscopy of imprinted materials. (a) MIP; (b) and (c) top surface and (d) cross-section of PMMA MIP. | ||
Semi-covalently imprinted polymer and its corresponding control were evaluated with respect to their ability to bind the template molecule, BPA, in aqueous solutions in equilibrium conditions. The binding affinity of BPA by NIP and semi-covalent MIP in aqueous solutions was tested in the range of 5–50 μM.
Fig. 3(a) shows the binding isotherms for NIP and MIP. As can be seen, the equilibrium binding of BPA increases with the initial concentration of the analyte and higher adsorption ability by the MIP is present across the whole concentration range.
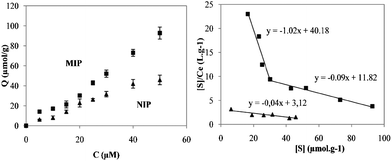 | ||
| Fig. 3 (a) BPA binding isotherms for NIP and MIP; (b) Scatchard analysis plots for both NIP and MIP. | ||
This is usually indicative of the presence of affinity binding sites created by the molecular imprinting process. Scatchard analysis was carried out to evaluate the binding properties of the semi-covalently imprinted polymer. Fig. 3(b) illustrates the Scatchard plots for BPA adsorption in MIP and NIP. As it can be seen, two straight lines could be wide-ranging withdrawn for the MIP. This result strongly suggests different binding sites affinity, very common in molecularly imprinted polymers.22 Although the copolymer was prepared by the semi-covalent approach, with the template and the monomer covalently bound, the analyte rebinding occurs by non-covalent interactions. From the straight area in the range of 16–30 μmol g−1, an affinity constant for the high-affinity binding sites MIP was determined to be 1.0 × 105 M−1 and an apparent maximum binding capacity of 39.2 μmol g−1 was attained. In the low affinity range, the association constants for MIP and NIP were, respectively, 0.9 × 105 M−1 and 0.4 × 105 M−1, whilst the maximum binding capacities were calculated to be 133.6 μmol g−1 and 74.5 μmol g−1. The results, presented in Table 2, show that the imprinted polymer possesses an overall higher affinity and binding capacity for the template molecule in the aqueous environment than the control polymer.
| Polymer | High-affinity sites | Low-affinity sites | ||
|---|---|---|---|---|
| K a × 105 (M−1) | B max (μmol g−1) | K a × 105 (M−1) | B max (μmol g−1) | |
| MIP | 1.0 | 39.2 | 0.9 | 133.6 |
| NIP | — | — | 0.4 | 74.5 |
The selectivity of MIP in aqueous solutions was assessed by evaluation of its capability to bind PRO and EE in comparison with BPA. The ability of imprinted polymers to selectively adsorb the template molecule in an aqueous environment is one of the most challenging features of MIPs and much attention is being focused on this topic.23Fig. 4 illustrates the data obtained for the selectivity experiments in aqueous solutions for both NIP and MIP, concerning the maximum adsorption capacities. Results show that NIP binds BPA and progesterone to the same extent, whereas the imprinted polymer binds progesterone to a much lower degree. This occurs because within a MIP, the functional groups are organized as a system of affinity binding sites, with a structure dependent upon the complementary affinity introduced at the imprinting stage, by the template molecule, BPA. In the NIP the functional groups of MAA have a comparatively random distribution, yielding different binding characteristics.24
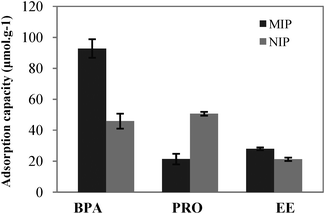 | ||
| Fig. 4 Maximum adsorption capacities for BPA, PRO and EE for both NIP and MIP. | ||
The imprinted polymer with molecular recognition to BPA, synthesized using a supercritical mixture of CO2 and acetonitrile, shows an adsorption capacity for the template that corresponds to 4.3 and 3.3 times the maximum adsorption of PRO and EE, respectively, which shows a water-compatible performance.
The feasibility of enhancing the analyte adsorption capacity by a membrane structure was evaluated by preparing a semi-covalently molecularly imprinted supported membrane by scCO2-assisted phase-inversion. Fig. 5 shows the BPA adsorbed by the imprinted and non-imprinted hybrid membranes in the dynamic binding experiments.
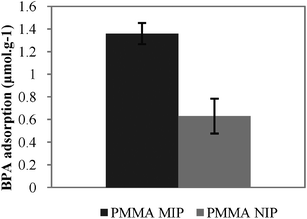 | ||
| Fig. 5 BPA adsorbed by the hybrid membranes in the dynamic binding experiments. | ||
Results show that with the incorporation of 30 wt% of BPA-imprinted polymer particles BPA imprinted polymer within the membrane (PMMA MIP) leads to a maximum adsorption of BPA (1.36 μmol g−1) which is around two-fold that of the maximum adsorbed by PMMA NIP (0.63 μmol g−1). This performance could be further tuned by controlling the amount of MIP particles incorporated in the membrane.
Conclusions
The work herein reported shows that supercritical fluid technology is a viable alternative to prepare molecularly imprinted polymeric materials using the semi-covalent approach. The synthesized polymer with molecular recognition to BPA showed a significant selectivity in aqueous solutions. MIP adsorbs higher amounts of BPA than the corresponding NIP material, whereas the adsorption capacity for PRO and EE remains low.The results demonstrate the feasibility of preparing semi-covalent MIPs capable of keeping their molecular recognition in aqueous media, using supercritical fluid technology, which can be a valuable alternative towards the synthesis of these MIPs in water. In addition, the incorporation of imprinted polymers in membranes opens up the possibility of increasing molecular affinity of porous structures to target molecules combining semi-covalent molecular imprinting and supercritical-assisted phase inversion.
Acknowledgements
The authors would like to thank Fundação para a Ciência e Tecnologia (FCT-Lisbon) for financial support through projects PTDC/QUI-QUI/102460/2008 and PTDC/CTM/70513/2006, and doctoral grant SFRH/BD/31085/2006 (M.S.S.) and grant PEst-C/EQB/LA0006/2011. V.D.B.B is supported by the MIT-Portugal Program (Bioengineering Systems Focus Area).References
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O'Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106 CrossRef CAS.
- J. Svenson and I. A. Nicholls, Anal. Chim. Acta, 2001, 435, 19 CrossRef CAS.
- C. Baggiani, G. Giraudi, F. Trotta, C. Giovannoli and A. Vanni, Talanta, 2000, 51, 71 CrossRef CAS.
- C. Lingxin, X. Shoufang and L. Jinhua, Chem. Soc. Rev., 2011, 40, 2922 RSC.
- C. Baggiani, P. Baravalle, L. Anfossi and C. Tozzi, Anal. Chim. Acta, 2005, 542, 125 CrossRef CAS.
- X. Shen and L. Ye, Chem. Commun., 2011, 47, 10359 RSC.
- S. Wei and B. Mizaikoff, J. Sep. Sci., 2007, 30, 1794 CrossRef CAS.
- C. Yu and K. Mosbach, J. Mol. Recognit., 1998, 11, 69 CrossRef CAS.
- M. Soares da Silva, E. R. Vão, M. Temtem, L. Mafra, J. Caldeira, A. Aguiar-Ricardo and T. Casimiro, Biosens. Bioelectron., 2010, 25, 1742 CrossRef CAS.
- A. R. C. Duarte, T. Casimiro, A. Aguiar-Ricardo, A. L. Simplício and C. M. M. Duarte, J. Supercrit. Fluids, 2006, 39, 102 CrossRef CAS.
- M. Soares da Silva, F. L. Nobrega, A. Aguiar-Ricardo, E. J. Cabrita and T. Casimiro, J. Supercrit. Fluids, 2011, 58, 150 CrossRef CAS.
- M. Soares da Silva, R. Viveiros, P. I. Morgado, A. Aguiar-Ricardo, I. J. Correia and T. Casimiro, Int. J. Pharm., 2011, 461, 61 CrossRef.
- M. Soares da Silva, R. Viveiros, M. B. Coelho, A. Aguiar-Ricardo and T. Casimiro, Chem. Eng. Sci., 2012, 68, 94 CrossRef CAS.
- E. Caroa, N. Masqué, R. M. Marcé, F. Borrulla, P. A. G. Cormack and D. C. Sherrington, J. Chromatogr., A, 2002, 963, 169 CrossRef.
- E. Burridge, Eur. Chem. News, 2003, 17, 14 Search PubMed.
- S. H. Dairkee, J. Seok, S. Champion, A. Sayeed, M. Mindrinos, W. Xiao, R. W. Davis and W. H. Goodson, Cancer Res., 2008, 68, 2076 CrossRef CAS.
- I. A. Lang, T. S. Galloway, A. Scarlett, W. E. Henley, M. Depledge, R. B. Wallace and D. Melzer, JAMA, J. Am. Med. Assoc., 2008, 300, 1303 CrossRef CAS.
- T. Casimiro, A. M. Banet-Osuna, M. Nunes da Ponte and A. Aguiar-Ricardo, Eur. Polym. J., 2005, 41, 1947 CrossRef CAS.
- T. Kobayashi, S. S. Leong and Q. J. Zhang, J. Appl. Polym. Sci., 2008, 108, 757 CrossRef CAS.
- T. Ikegami, T. Mukawa, H. Nariai and T. Takeuchi, Anal. Chim. Acta, 2004, 504, 131 CrossRef CAS.
- A. F. Abdel-Magid, J. H. Cohen, C. A. Maryanoff, R. D. Shah, F. J. Villani and F. Zhang, Tetrahedron Lett., 1998, 39, 3391 CrossRef CAS.
- C. J. Allender, K. R. Brain and C. M. Heard, Chirality, 1997, 9, 233 CrossRef CAS.
- C. Lingxin, X. Shoufang and L. Jinhua, Chem. Soc. Rev., 2011, 40, 2922 RSC.
- H. Hiratani and C. Alvarez-Lorenzo, J. Controlled Release, 2002, 83, 223 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |