Ionic liquid 1,3-disulfonic acid imidazolium hydrogen sulfate: a novel and highly efficient catalyst for the preparation of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols†
Abdolkarim
Zare
*a,
Tahereh
Yousofi
a and
Ahmad Reza
Moosavi-Zare
*b
aDepartment of Chemistry, Payame Noor University, PO BOX 19395-4697, Tehran, Iran. E-mail: abdolkarimzare@yahoo.com; Fax: +98 771 5559489; Tel: +98 771 5559488
bFaculty of Chemistry, Bu-Ali Sina University, Hamedan, 6517838683, Iran. E-mail: moosavizare@yahoo.com; Fax: +98 811 8258407; Tel: +98 771 8282807
First published on 11th July 2012
Abstract
In this work, a novel ionic liquid, 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4}, with a Brønsted acidic property, is synthesized, and used as a highly efficient, green, recyclable and homogeneous catalyst for the preparation of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols under solvent-free and relatively mild conditions. The one-pot multi-component condensation of β-naphthol with arylaldehydes and alkyl carbamates or amides affords the title compounds in high yields and in short reaction times.
Ionic liquids (ILs) have attracted increasing interest from chemists in the last few decades, because of their unique properties including non-flammability, non-volatility, wide liquid-state temperature range, high thermal and chemical stability, large electrochemical window and favorable salvation behavior.1–3 These compounds have been extensively used in electrochemistry, spectroscopy, extraction and separation processes,2 and also as a solvent, catalyst and reagent in organic synthesis.1–15 Brønsted acidic ILs are an important class of these salts, which have offered the possibility for development of environmentally friendly acidic catalysts for organic transformations, due to the combined advantages of liquid and solid acids, their operational simplicity, efficacy and selectivity coupled with their green natures.9–15
1-Carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols are of importance as they can be easily hydrolyzed to 1-aminoalkyl-2-naphthol derivatives, which are biologically interesting compounds. 1-Aminoalkyl-2-naphthols have been frequently used as hypotensive and bradycardiac agents.16,17 1-Amidoalkyl-2-naphthols can be also converted to 1,3-oxazine derivatives.18 Several pharmaceutical properties have been reported for 1,3-oxazines, e.g., antibiotic,19 antitumor,20 and analgesic activities.21
The one-pot multi-component condensation reaction between β-naphthol, aromatic aldehydes and alkyl carbamates22–25 or amides15,26–35 has been used as a practical synthetic route towards 1-carbamatoalkyl-2-naphthol22–25 and 1-amidoalkyl-2-naphthol15,26–35 derivatives. Although some catalysts and methods for the synthesis of the title compounds are known, newer catalysts and methods continue to attract attention for their differences with the others in terms of generality and effectiveness. Furthermore, most of the reported methods for the synthesis of the 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols are associated with one or more of the following disadvantages: (i) the use of large amount of catalyst, (ii) the use of expensive and toxic catalysts, (iii) non-availability of the catalyst or its starting materials, (iv) poor agreement with the green chemistry protocols, (v) moderate yield, (vi) long reaction time, (vii) harsh reaction conditions, and especially (viii) poor generality (in most of the reported procedures, the synthesis of one type of the title compounds has been reported).
Having the above points in mind, we report here the synthesis of a new Brønsted acidic ionic liquid, 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4}, from available and inexpensive starting materials (Scheme 1),36 and its application as a highly efficient, homogeneous, recyclable and green catalyst for the preparation of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols. The one-pot multi-component condensation of β-naphthol with arylaldehydes and alkyl carbamates or amides under solvent-free and relatively mild reaction conditions affords the compounds mentioned in Scheme 2.37 It is noteworthy that our method has none of the above-mentioned drawbacks.
![The synthesis of 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4}.](/image/article/2012/RA/c2ra20679j/c2ra20679j-s1.gif) | ||
| Scheme 1 The synthesis of 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4}. | ||
![The synthesis of 1-carbamatoalkyl-2-naphthols (1a–g) and 1-amidoalkyl-2-naphthols (2a–i) using [Dsim]HSO4.](/image/article/2012/RA/c2ra20679j/c2ra20679j-s2.gif) | ||
| Scheme 2 The synthesis of 1-carbamatoalkyl-2-naphthols (1a–g) and 1-amidoalkyl-2-naphthols (2a–i) using [Dsim]HSO4. | ||
Initially, the ionic liquid 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4} was synthesized (Scheme 1) and identified by IR, 1H NMR, 13C NMR and mass spectra.36 Furthermore, to confirm that 1,3-disulfonic acid imidazolium chloride {[Dsim]Cl} has been completely converted to our catalyst [Dsim]HSO4, a solution of AgNO3 in distilled water was added to a solution of the catalyst in distilled water. The absence of AgCl precipitate indicated complete conversion of the [Dsim]Cl to [Dsim]HSO4.9,11
In another study, thermal gravimetric (TG) and derivative thermal gravimetric (DTG) analysis of [Dsim]HSO4 was investigated at range of 25 to 600 °C, with a temperature increase rate of 10 °C min−1 in a nitrogen atmosphere (Fig. 1). The TG and DTG data of the catalyst showed two major weight losses; the first was observed after 350 °C, and the second appears after 540 °C. Therefore, the molecular decomposition of the ionic liquid occurred after 350 °C. The weight losses, which corresponded to about 24%, 8%, 13% and 24% of the ionic liquid weight, can be related to the loss of SO3, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, CH3CN and SO3, respectively.
CH2, CH3CN and SO3, respectively.
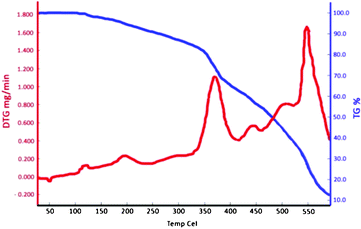 | ||
| Fig. 1 The TG and DTG diagrams of the ionic liquid. | ||
After characterization of the ionic liquid, its efficacy to catalyze the preparation of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols was examined. For this purpose, as a model reaction, effect of different molar ratios of [Dsim]HSO4 and temperature on the one-pot multi-component condensation of β-naphthol with benzaldehyde and methyl carbamate in the absence of solvent was studied. The results showed that 5 mol% of the catalyst was sufficient to promote the reaction efficiently at 80 °C; in these conditions, the product was obtained in 98% yield after 11 min.
In order to assess the efficiency and the generality of the catalyst, β-naphthol was reacted with different arylaldehydes (including benzaldehydes and arylaldehydes possessing electron-withdrawing and electron-releasing substituents as well as halogens) and alkyl carbamates (methyl and benzyl carbamates) under the optimal reaction conditions. The results are displayed in Table 1. Ascan be seen in Table 1, all reactions proceeded effectively and afforded the corresponding 1-carbamatoalkyl-2-naphthol derivatives in excellent yields in short reaction times (Table 1, compounds 1a–g). Interestingly, our catalyst, [Dsim]HSO4, worked well when amides instead of alkyl carbamates were used; in these conditions, 1-amidoalkyl-2-naphthols were obtained in high yields and in short reaction times from β-naphthol, various arylaldehydes (including benzaldehydes and arylaldehydes bearing electron-withdrawing and electron-releasing substituents as well as halogens) and various amide derivatives (such as acetamide, acrylamide and benzamide) (Table 1, compounds 2a–i). Thus, the catalyst was highly efficient and general for the synthesis of the mentioned compounds.
| Ar | RO or R′ | Product | Timea/Yieldb | M.p. (°C) (Lit.) |
|---|---|---|---|---|
| a Reaction time in min. b Isolated yield in %. | ||||
| C6H5 | CH3O | 1a | 11/98 | 220–222 |
| (222–224)23 | ||||
| 3-NO2C6H5 | CH3O | 1b | 7/98 | 249–251 |
| (253–255)23 | ||||
| 4-NO2C6H5 | CH3O | 1c | 8/98 | 203–205 |
| (200–202)23 | ||||
| 4-ClC6H5 | CH3O | 1d | 15/96 | 201–203 |
| (198–200)25 | ||||
| 4-BrC6H5 | CH3O | 1e | 17/98 | 172–174 |
| C6H5 | C6H5CH2O | 1f | 14/95 | 177–179 |
| (179–180)25 | ||||
| 3-CH3OC6H5 | C6H5CH2O | 1g | 25/91 | 181–183 |
| (182–184)25 | ||||
| C6H5 | CH3 | 2a | 9/98 | 239–240 |
| (239–240)26 | ||||
| 4-NO2C6H5 | CH3 | 2b | 10/97 | 248–250 |
| (247–249)26 | ||||
| 4-CH3OC6H5 | CH3 | 2c | 25/98 | 182–184 |
| (184–186)27 | ||||
| 4-BrC6H5 | CH3 | 2d | 14/96 | 226–228 |
| (228–230)28 | ||||
| 4-FC6H5 | CH3 | 2e | 12/97 | 206–208 |
| (206–208)26 | ||||
| 4-CH3C6H5 | CH2CH |
2f | 18/97 | 216–217 |
| (214–216)29 | ||||
| 4-NO2C6H5 | CH2CH |
2g | 10/98 | 218–220 |
| (223–225)29 | ||||
| 3-NO2C6H5 | C6H5 | 2h | 20/97 | 215–217 |
| (214–216)26 | ||||
| 4-CH3OC6H5 | C6H5 | 2i | 35/95 | 202–204 |
| (208–210)30 | ||||
To compare the efficiency of our catalyst with the reported catalysts for the synthesis of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols, we have tabulated the results of these catalysts to promote the synthesis of compounds 1a and 2a, in Table 2. As shown in Table 2, our catalyst is superior to the previously reported catalysts in terms of reaction temperature, reaction time and yield. Furthermore, in our presented work, the preparation of both 1-carbamatoalkyl-2-naphthol and 1-amidoalkyl-2-naphthol derivatives have been achieved.
| Catalyst, reaction temperature (°C) | Compound 1a | Compound 2a | ||
|---|---|---|---|---|
| Time (min) | Yield (%) | Time (min) | Yield (%) | |
| a Our catalyst. b In this paper, the synthesis of 1-carbamatoalkyl-2-naphthols have not been reported. c In this paper, the synthesis of 1-amidoalkyl-2-naphthols have not been reported. | ||||
| [Dsim]HSO4, 80a | 11 | 98 | 9 | 98 |
| 1,3-Disulfonic acid imidazolium chloride, 12015 | —b | —b | 1 | 95 |
| 4-(1-Imidazolium) butanesulfonate, 8022 | 120 | 78 | 120 | 80 |
| Preyssler nanoparticles/SiO2, 9023 | 3 | 84 | —c | —c |
| NaHSO4/SiO2, 10025 | 3.5 | 81 | —c | —c |
| H3PMo12O40.xH2O/SiO2, 12026 | —b | —b | 15 | 91 |
| Ce(SO4)2, 8527 | —b | —b | 2160 | 72 |
| Montmorillonite K10 clay, 12528 | —b | —b | 90 | 89 |
| PEG based dicationic acidic ionic liquid, 8029 | —b | —b | 5 | 91 |
| Copper p-toluene sulfonate, 8030 | —b | —b | 90 | 94 |
| HClO4/SiO2, 11031 | —b | —b | 40 | 89 |
| Sulfamic acid (ultrasound), 3032 | —b | —b | 15 | 89 |
| Silphox, 12033 | —b | —b | 30 | 92 |
| Silphos, 12033 | —b | —b | 20 | 93 |
| [Bmim]Br (microwave), 13034 | —b | —b | 25 | 94 |
| Silica chloride (ultrasound), 3035 | —b | —b | 9 | 95 |
To increase the catalyst worth, its recyclability was tested upon the synthesis of compound 1a. After completion of the reaction, the product was extracted by ethyl acetate (the product is soluble in ethyl acetate, but [Dsim]HSO4 is not soluble in this solvent), and the remaining catalyst was used for the next run of the reaction. The catalytic activity of [Dsim]HSO4 was restored within the limits of the experimental errors for four successive runs (see Table 3).
In a plausible mechanism (Scheme 3), which is supported by the literature;15,22,25,29–31 at first, β-naphthol attacks the activated aldehyde by [Dsim]HSO4 to give intermediate I. Afterward, ortho-quinone methide (o-QM) is produced by removing one molecule of H2O from I. Subsequently, alkyl carbamate or amide reacts with o-QM as a Michael acceptor to afford 1-carbamatoalkyl-2-naphthols or 1-amidoalkyl-2-naphthols. Because of the specific structure of the catalyst, it can form several hydrogenic bonds with the starting materials by its Brønsted acidic and Lewis basic sites. Due to the formation of these hydrogenic bonds, the starting materials could be positioned in a suitable state to react with each other. On the other hand, 1,3-disulfonic acid imidazolium hydrogen sulfate is more acidic than 1,3-disulfonic acid imidazolium chloride (pH of a 0.02 M solution of [Dsim]HSO4 and [Dsim]Cl were 1.8 and 2.3, respectively); thus, the former ionic liquid can activate aldehydes and o-QM better than the latter. For these reasons, [Dsim]HSO4 is a more successful catalyst than [Dsim]Cl for this reaction.
![The proposed mechanism for the preparation of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols using [Dsim]HSO4.](/image/article/2012/RA/c2ra20679j/c2ra20679j-s3.gif) | ||
| Scheme 3 The proposed mechanism for the preparation of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols using [Dsim]HSO4. | ||
Conclusions
In summery, we have prepared and applied a novel ionic liquid, namely 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4}, as a highly efficient, homogeneous, recyclable and green catalyst for the synthesis of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols from β-naphthol, arylaldehydes and alkyl carbamates or amides. This method has solved most of the drawbacks accompanied with previously reported methods.Acknowledgements
The authors gratefully acknowledge financial support of this work by Research Council of Payame Noor University.References
- P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef CAS.
- J. Pavlinac, M. Zupan, K. K. Laali and S. Stavber, Tetrahedron, 2009, 65, 5625 CrossRef CAS.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal. A: Gen., 2010, 373, 1 CrossRef CAS.
- C. Meyer, V. Hager, W. Schwieger and P. Wasserscheid, J. Catal., 2012, 292, 157 CrossRef CAS.
- Y. J. Kim and R. S. Varma, Tetrahedron Lett., 2005, 46, 1467 CrossRef CAS.
- A. Zare, A. Hasaninejad, A. Khalafi-Nezhad, A. R. Moosavi-Zare, M. H. Beyzavi, F. Khedri, F. Asadi, N. Hayati and A. Asifi, J. Iran. Chem. Soc., 2010, 7, 461 CrossRef CAS.
- A. Hasaninejad, A. Zare, M. Shekouhy and J. Ameri Rad, J. Comb. Chem., 2010, 12, 844 CrossRef CAS.
- H. Valizadeh, M. Amiri and F. Hosseinzadeh, Dyes Pigm., 2012, 92, 1308 CrossRef CAS.
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, A. Zare, H. G. Kruger, Z. Asgari, V. Khakyzadeh and M. Kazem-Rostami, J. Org. Chem., 2012, 77, 3640 CrossRef CAS.
- A. Zare, A. R. Moosavi-Zare, M. Merajoddin, M. A. Zolfigol, T. Hekmat-Zadeh, A. Hasaninejad, A. Khazaei, M. Mokhlesi, V. Khakyzadeh, F. Derakhshan-Panah, M. H. Beyzavi, E. Rostami, A. Arghoon and R. Roohandeh, J. Mol. Liq., 2012, 167, 69 CrossRef CAS.
- N. G. Khaligh, J. Mol. Catal. A: Chem., 2011, 349, 63 CrossRef CAS.
- X. Li, Y. Gu, Y. Yang, H. Song and S. Yao, Adv. Mater. Res., 2012, 396–398, 1969 CAS.
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare and A. Zare, J. Iran. Chem. Soc., 2010, 7, 646 CrossRef CAS.
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare and A. Zare, Org. Prep. Proced. Int., 2010, 42, 95 CrossRef CAS.
- M. A. Zolfigol, A. Khazaei, A. R. Moosavi-Zare, A. Zare and V. Khakyzadeh, Appl. Catal. A: Gen., 2011, 400, 70 CrossRef CAS.
- H. R. Shaterian, H. Yarahmadi and M. Ghashang, Bioorg. Med. Chem. Lett., 2008, 18, 788 CrossRef CAS.
- A. Y. Shen, C. T. Tsai and C. L. Chen, Eur. J. Med. Chem., 1999, 34, 877 CrossRef CAS.
- M. Damodiran, N. P. Selvam and P. T. Perumal, Tetrahedron Lett., 2009, 50, 5474 CrossRef CAS.
- Y. Kusakabe, J. Nagatsu, M. Shibuya, O. Kawaguchi, C. Hirose and S. Shirato, J. Antibiot., 1972, 25, 44 CrossRef CAS.
- S. Renullard, L. I. Rebhun, G. A. Havic and S. M. Kupchan, Science, 1975, 189, 1002 Search PubMed.
- G. Y. Lesher and A. R. Surrey, J. Am. Chem. Soc., 1955, 77, 636 CrossRef CAS.
- D. Kundu, A. Majee and A. Hajra, Catal. Commun., 2010, 11, 1157 CrossRef CAS.
- M. M. Heravi, N. Tavakoli-Hoseini and F. F. Bamoharram, Green Chem. Lett. Rev., 2010, 3, 263 CrossRef CAS.
- H. R. Shaterian, A. Hosseinian and M. Ghashang, Chin. J. Chem., 2009, 27, 821 CrossRef CAS.
- H. R. Shaterian, A. Hosseinian and M. Ghashang, Tetrahedron Lett., 2008, 49, 5804 CrossRef CAS.
- A. Zare, A. Hasaninejad, E. Rostami, A. R. Moosavi-Zare, N. Pishahang, M. Roshankar, F. Khedri and M. Khedri, E-J. Chem., 2010, 7, 1162 CrossRef CAS.
- N. P. Selvam and P. T. Perumal, Tetrahedron Lett., 2006, 47, 7481 CrossRef CAS.
- S. Kantevari, S. V. N. Vuppalapati and L. Nagarapu, Catal. Commun., 2007, 8, 1857 CrossRef CAS.
- J. Luo and Q. Zhang, Monatsh. Chem., 2011, 142, 923 CrossRef CAS.
- M. Wang and Y. Liang, Monatsh. Chem., 2011, 142, 153 CrossRef CAS.
- H. R. Shaterian, H. Yarahmadi and M. Ghashang, Tetrahedron, 2008, 64, 1263 CrossRef CAS.
- S. B. Patil, P. R. Singh, M. P. Surpur and S. D. Samant, Ultrason. Sonochem., 2007, 14, 515 CrossRef CAS.
- A. Zare, Org. Prep. Proced. Int., 2012, 44, 82 CrossRef CAS.
- A. Zare, A. Hasaninejad, A. Salimi Beni, A. R. Moosavi-Zare, M. Merajoddin, E. Kamali, M. Akbari-Seddigh and Z. Parsaee, Sci. Iran., Trans. C, 2011, 18, 433 CAS.
- B. Datta and M. A. Pasha, Ultrason. Sonochem., 2011, 18, 624 CrossRef CAS.
- Procedure for the preparation of ionic liquid 1,3-disulfonic acid imidazolium hydrogen sulfate {[Dsim]HSO4} (Scheme 1): to a round-bottomed flask (100 mL) containing imidazole (0.340 g, 5 mmol) in dry CH2Cl2 (50 mL), was added chlorosulfonic acid (1.1885 g, 10.2 mmol) dropwise over a period of 20 min at room temperature. After the addition was completed, the reaction mixture was stirred for 12 h under pressure of nitrogen gas, stood for 5 min, and the CH2Cl2 was decanted. The residue was washed with dry CH2Cl2 (3×50 mL) and dried under vacuum to give 1,3-disulfonic acid imidazolium chloride {[Dsim]Cl} as a viscous pale yellow oil in 95% yield.15 Then, sulfuric acid (99.99%) (0.49 g, 5 mmol) was added dropwise to [Dsim]Cl (1.63 g, 5 mmol) over a period of 5 min at room temperature under pressure of nitrogen gas (to remove HCl produced during the reaction). The resulting mixture was stirred for 24 h at 60 °C under a continuous flow of nitrogen gas to give [Dsim]HSO4 as a viscous yellow oil in 99% yield. IR (Nujol): 624, 1031, 1053, 1085, 1285, 1324, 3100–3400 cm−1; 1H NMR (300 MHz, DMSO-d6): δ 7.22 (s, 2H), 8.44 (s, 1H), 11.95 (s, 1H), 13.55 (s, 2H); 13C NMR (75 MHz, DMSO-d6): δ 119.5, 134.0; MS (m/z): 326 (M+).
- General procedure for the synthesis of 1-carbamatoalkyl-2-naphthols and 1-amidoalkyl-2-naphthols (Scheme 2): to a mixture of β-naphthol (0.288 g, 2 mmol), arylaldehyde (2 mmol) and alkyl carbamate or amide (2.4 mmol) in a test tube, was added [Dsim]HSO4 (0.033 g, 0.1 mmol), and the resulting mixture was firstly stirred magnetically, and after solidification of the reaction mixture with a small rod, at 80 °C. After completion of the reaction, as monitored by TLC, the reaction mixture was cooled to room temperature, H2O (5 mL) was added, stirred for 3 min and filtered. The solid residue was recrystallized from hot EtOH (95%) to give the pure product.
Footnote |
| † Electronic Supplementary Information (ESI) available: General experimental details for the starting materials and instruments, and selected spectral data of the products. See DOI: 10.1039/c2ra20679j/ |
| This journal is © The Royal Society of Chemistry 2012 |