Synthesis of Au and Ag nanoparticles with alkylselenocyanates†
Oksana
Zaluzhna
a,
Chris
Zangmeister
b and
YuYe J.
Tong
*a
aDepartment of Chemistry, Georgetown University, 37th & O Streets, NW, Washington DC 20057, USA. E-mail: yyt@georgetown. edu
bNational Institute of Standards and Technology, 100 Bureau Drive, Gaithersburg, MD 20899, USA
First published on 18th June 2012
Abstract
We report the first successful synthesis of Au and Ag nanoparticles (NPs) with alkylselenocyanates as the source of protecting ligands. The resultant NPs were characterized to better understand the interfacial chemistry between the metal core and ligand by comparing and contrasting with the corresponding 2D self-assembled monolayers (SAMs).
Discovery of the thiolate-protected Au NP synthesis by the Brust and Schiffrin method (BSM)1,2 opened the door for extensive research in this field with a variety of ligands and metals.3–10 Although the mechanism for the formation of thiolate-protected metal NPs (where the ligand is anchored by S) is better understood,11–14 similar investigations on other heavy chalcogens (i.e., Se and Te)-anchored ligand-protected metal NPs are rare.15,16 According to quantum calculations, heavy chalcogen anchoring may have promising electronic properties, such as better conductance than the prevalent anchoring element S.17 While a few groups have investigated self-assembled monolayers (SAMs) with dissociative dichalcogenide (i.e., R2Se2 and R2Te2) adsorption on Au and Ag substrates, in which the broken dichalcogenide bond leads to Se or Te anchoring,18–22 studies on the corresponding Au and Ag NPs are scarce.6,23–25 The majority of these studies have been performed with only one type of molecular precursor, dialkyl dichalcogenides, as the starting ligands, since heavy chalcogenols are more air-sensitive26 than their light chalcogen cousin, thiol. To further open this important and promising field of research on anchoring chemistry of heavy chalcogens, developing good synthetic alternatives with different heavy chalcogen-containing starting ligands is highly desirable.
Ciszek et al. have recently used organic thiocyanates as alternative precursors for forming 2 dimensional (2D) SAMs primarily on Au but also on Ag and Pt and determined that thiocyanates assemblies resemble thiolate SAMs.27,28 It was demonstrated that the surface coverage remains better for widely-used thiols-generated SAMs than for thiocyanates-generated SAMs, yet thiocyanate-generated SAMs showed superior stability on the surfaces and avoided undesired polymerization processes.27,29,30 Among others, the main challenge in characterizing the thiocyanate-generated SAMs was to identify and quantify the possible presence of (CN) species. The authors argued that the surface morphology played a significant role in the expulsion of (CN)ads from metal surfaces, i.e., SAMs on evaporated Au surface had less residual nitrogen than the stripped template.28,29
Inspired by the work discussed in the previous paragraph, we used the same type of ligand to synthesize metal (Au and Ag) NPs via the BSM and reported the successful outcome in this Communication. To our best knowledge, the only work that has dealt with organo selenocyanates as a starting precursor for generating potential protecting ligands for NPs was the work by Lim et al. but in a very different context.31 They investigated the adsorption of nitrophenyl selenocyanate on the surface of pre-formed Au NPs stabilized by citrate via ligand exchange and compared it with benzeneselenol.31 Despite the lack of TEM image evidence, the authors assumed that the size of the colloidal ensemble remained unchanged following the ligand exchange, i.e., 15 nm spherical NPs.31 In this Communication, we focus on using the general BSM to synthesize metal (Au and Ag) NPs with alkylselenocyanates as the starting molecular precursor for the final stabilizing ligand and to compare and contrast the formation chemistry of the 3D SAM analogues to that of the corresponding 2D SAMs.
Several alkylselenocyanates ligands, such as C6H13SeCN, C8H17SeCN, and C12H25SeCN, were synthesized32 (see the Electronic Supporting Information, ESI†) and characterized via1H NMR (Fig. S1), 13C NMR (Fig. S2), IR (Fig. S3), Raman (Fig. S4), and 77Se NMR (Fig. S5). For the purpose of this Communication, only dodecylselenocyanate, i.e., C12H25SeCN or briefly C12SeCN, as the starting ligand will be discussed as a representative case to illustrate our main findings. Both one-phase (1p) and two-phase (2p) BSMs were employed to synthesize the Au NPs. While the 2p BSM was successful in producing Au NPs (Fig. S6, ESI†), the 1p BSM produced more homogenous NPs with C12SeCN. Therefore, the latter will be the focus of the study and discussion here. For details on other syntheses, readers are referred to the ESI,† specifically Fig. S7 for the results of other 1p syntheses.
TEM, UV-Vis, and size distribution data are presented in Fig. 1 for the C12Se-protected Au NPs synthesized by the 1p BSM (we will omit CN in naming NPs below because CN was a very minor species there, vide infra). The resultant NPs were homogenous, with an average particle size of 2.0 ± 0.3 nm (Fig. 1a), and exhibited a narrow size distribution (Fig. 1b). The UV-Vis did not display a clear surface plasmon resonance peak around 500–520 nm due to the small size of NPs (Fig. 1c).33
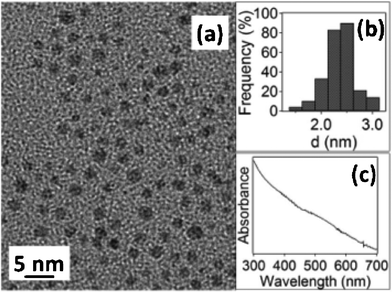 | ||
| Fig. 1 (a) TEM image, (b) size distribution histogram, and (c) UV-Vis spectrum of the 2.0 ± 0.3 nm C12Se–Au NPs. | ||
The obtained NPs were characterized via1H NMR, IR, and XPS. Fig. 2 displays the 1H NMR results for the NPs (Fig. 2a) and the free ligand (Fig. 2b). The triplet peak corresponding to the protons in –H2C–SeCN at 2.09 ppm in the free ligand (Fig. 2b) disappeared in the NP spectrum (Fig. 2a), indicating that the organic moiety is bound to the metal surface via Se. Also, note that the broadening and down-field shift of the alkyl chain peaks at 1.0 to 1.5 ppm is indicative of the ligand bound to the NPs.34 Unfortunately, 13C NMR lacked the sensitivity to determine the presence of C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N species within the NPs sample, as illustrated by the difficulty to observe the CN peak even in the free RSe–CN ligand (Fig. S2b†).
N species within the NPs sample, as illustrated by the difficulty to observe the CN peak even in the free RSe–CN ligand (Fig. S2b†).
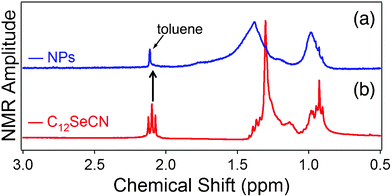 | ||
| Fig. 2 1H NMR spectra of (a) C12Se–Au NPs, and (b) pure C12SeCN ligand. | ||
The IR characterization was performed to determine whether the CN species were present on the Au NPs surface. As shown in Fig. 3, the cyanide peak at 2150 cm−1 (Fig. 3b) that was present in the free ligand disappeared completely in the NPs sample (Fig. 3a). That is, within the detection limit of IR, no residual CN was observed after the formation of Au NPs. The disappearance of the Se–C (trans) vibrational band at 630 cm−1 in the Au NPs sample is also indicative of the formation of Se–Au bond. The characteristic triplet peaks of methylene/methyl stretching frequencies appear at νs(CH2)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) 2852 cm−1, νas(CH2) = 2924 cm−1, and νas(CH3) = 2956 cm−1, respectively. These should be compared to the corresponding values of 2D SAMs:30 νs(CH2) = 2880 cm−1, νas(CH2) = 2935 cm−1, and νas(CH3) = 2965 cm−1. The smaller (red-shifted) frequencies observed on the Au NPs suggest that the alkylselenoate formed a more ordered monolayer structure on the NPs than on the Au(111) surface.35
2852 cm−1, νas(CH2) = 2924 cm−1, and νas(CH3) = 2956 cm−1, respectively. These should be compared to the corresponding values of 2D SAMs:30 νs(CH2) = 2880 cm−1, νas(CH2) = 2935 cm−1, and νas(CH3) = 2965 cm−1. The smaller (red-shifted) frequencies observed on the Au NPs suggest that the alkylselenoate formed a more ordered monolayer structure on the NPs than on the Au(111) surface.35
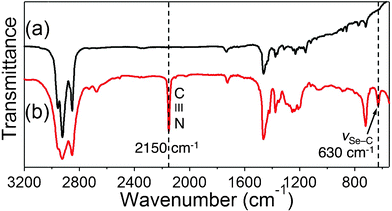 | ||
| Fig. 3 IR spectra of (a) C12SeCN–Au NPs, and (b) pure C12SeCN ligand. | ||
For their work on 2D SAMs, Ciszek et al. detected the presence of a very small amount of residual CN by using XPS and proposed the existence of two kinds of cyanides within the samples: the adsorbed cyanide (CNads) and an intermediate form absorbed on the surface during the etching of the metal (Au(CN)) intermediate species with vibrational IR band at ∼2160 cm−1,28 although their respective quantification was proved to be challenging. Choi et al. had also found it challenging to identify and quantify the presence of cyano species in their octylthiocyanate-generated SAMs on Au(111).29 They predicted that the CN bands could be more readily observed on the surface of Au via SERS, though the amount of CN was impossible to be determined.29,31 Our IR results also suggest that there is little, if any, cyanide species present in the NPs sample, since no observable CN vibrational peaks were present in the spectra.
XPS analysis, as a more sensitive tool of elemental analysis, was used to further analyze the ligand-metal characteristics (Fig. 4 and Fig. S8 in the ESI†). Fig. 4a displays the XPS spectra of Se 3d5/2 with a binding energy of 55.3 eV, which has been observed for the C12Se–Au NPs synthesized with dialkyl-diselenides as the starting ligand.15,24 The same binding energies for the Au 4f7/2 doublet (87.75 eV and 84.05 eV respectively, Fig. S8) were also observed. Altogether, these binding energies lead to the conclusion that the organic moiety is indeed bound to the Au surface via the chalcogen and the Au NPs synthesized with alkylselenocyanate and dialkyl-diselenide are essentially the same.
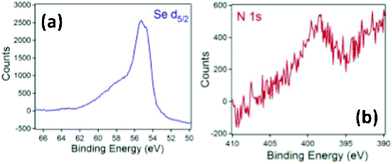 | ||
| Fig. 4 XPS of (a) Se d5/2, and (b) N 1s region in C12SeCN–Au NPs. | ||
Furthermore, XPS was used to investigate the existence of (CN) species within the Au NPs. As mentioned previously, Ciszek et al. proposed the existence of two types of cyanides within the SAMs samples, i.e., CNads on Au (with XPS binding energy of N 1s electron = 397.9 eV) and Au(CN) intermediate species (with XPS binding energy of N 1s = 399.5 eV).28 For our study, a weak and noisy N 1s XPS signal was observed at ∼399 eV in our samples (Fig. 4b) as one broad peak in the spectra. Unfortunately, the weak signal and peak broadening (Fig. 4b) on the C12Se–Au NPs sample made the differentiations of the two possible cyano species impossible. Based on these results, we can ascertain that the Au NPs synthesized with alkylselenocyanate are predominantly protected by alkylselenolate, while (CN) species exist in a trace amount that cannot be determined quantitatively.
Additionally, TGA was performed on the NPs and showed a two-step mass loss (Fig. S9†). The first step occurred at ∼180 °C of about 30% mass loss, and the second at ∼350 °C of about 13% mass loss. The first mass-loss temperature corresponded closely to the loss of the organic alkyl chain with Se as an anchoring element, as previously reported in the literature.24 The second mass loss, on the other hand, warrants more investigation, since the mass loss is too large to correspond to the cyano species.
We also carried out similar, but less extensive synthesis of Ag NPs with C12SeCN using 2p BMS. Despite being proved much more challenging to synthesize than Au NPs in terms of reproducibility of size homogeneity, Fig. 5 presents the TEM image, size distribution histogram, and UV-Vis spectrum of our best sample. While the size of the synthesized Ag NPs (2.9 ± 0.3 nm) is larger than that of the corresponding Au NPs (2.0 ± 0.3 nm), very similar IR (disappearance of the C–N and the Se–C stretching bands in the Ag NPs, Fig. S10) and TGA (the two-step desorption, Fig. S11) were observed. The vibrational frequencies of the methyl/methylene triplet were 2846 cm−1, 2916 cm−1, and 2954 cm−1, respectively. These frequencies are further red-shifted as compared to those on the Au NPs, suggesting that even more ordered monolayer was formed on the Ag NPs.35 All these data indicate that the ligand monolayer on the Ag NPs shares more structural and chemical similarity than difference with its counterpart on the Au NPs.
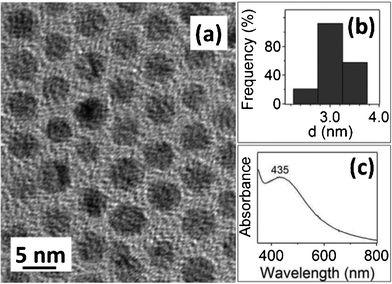 | ||
| Fig. 5 (a) TEM image, (b) size distribution histogram, and (c) UV-Vis spectrum of the 2.9 ± 0.3 nm C12Se–Ag NPs. | ||
Conclusions
In conclusion, alkylselenocyanates were successfully used for the first time as the starting ligands for synthesizing homogenous, ca. 2 to 3 nm sized, Au and Ag NPs by using the popular BSM, which opens a promising and general alternative to synthesizing organo-heavier-chalcognen-ligand protected metal NPs. The 1H NMR, IR, and XPS data all suggest that the ligands were bonded to the NP surface via Se. Strong evidence also indicates that the ligand monolayer is most likely alkylselenolate, the same as that on the Au NPs synthesized with dialkyl-diselenide and on Au(111), and the Au and Ag NPs most likely shared a very similar monolayer structure that was more ordered than the corresponding 2D SAMs. We believe that the reaction mechanism involving the starting alkylcyanates as proposed by Ciszek et al.27,28 may apply to the NPs synthesis. Briefly, the RSe–CN bond was broken during the synthesis. While RSe– acts as stabilizing ligand for the NPs, CN would form a Au/Ag(CN)2 complex and be washed away during post-synthesis purification process. However, the determination of CN species in the NPs was proved to be a task as challenging as in the 2D SAMs, due largely to the low sensitivity of available instruments. While IR failed to detect the presence of CN species, XPS showed a broad, weak, and noisy peak that most likely corresponded to the trace amount of cyanide present in the synthesized (Au) NPs. Analytical techniques with higher sensitivity are needed to fully characterize the residual CN.
Acknowledgements
This work was supported by grants from the NSF (CHE-0923910) and DOE (DE-FG02-07ER15895). The authors thank the UMD NISP Lab for use of its TEM facility.References
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- B. S. Zelakiewicz, G. C. Lica, M. L. Deacon and Y. Y. Tong, J. Am. Chem. Soc., 2004, 126, 10053–10058 CrossRef CAS.
- B. S. Zelakiewicz, T. Yonezawa and Y. Y. Tong, J. Am. Chem. Soc., 2004, 126, 8112–8113 CrossRef CAS.
- G. C. Lica, B. S. Zelakiewicz, M. Constantinescu and Y. Y. Tong, J. Phys. Chem. B, 2004, 108, 19896–19900 CrossRef CAS.
- Y. Li, L. C. Silverton, R. Haasch and Y. Y. Tong, Langmuir, 2008, 24, 7048–7053 CrossRef CAS.
- O. Zaluzhna, L. Brightful, T. C. Allison and Y. Y. J. Tong, Chem. Phys. Lett., 2011, 509, 148–151 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C.-J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2003, 104, 293–346.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS.
- P. J. G. Goulet and R. B. Lennox, J. Am. Chem. Soc., 2010, 132, 9582–9584 CrossRef CAS.
- Y. Li, O. Zaluzhna, B. Xu, Y. Gao, J. M. Modest and Y. Y. J. Tong, J. Am. Chem. Soc., 2011, 133, 2092–2095 CrossRef CAS.
- Y. Li, O. Zaluzhna and Y. Y. J. Tong, Chem. Commun., 2011, 47, 6033–6035 RSC.
- Y. Li, O. Zaluzhna and Y. Y. J. Tong, Langmuir, 2011, 27, 7366–7370 CrossRef CAS.
- O. Zaluzhna, Y. Li, C. Zangmeister, T. C. Allison and Y. Y. J. Tong, Chem. Commun., 2012, 48, 362–364 RSC.
- Y. Li, O. Zaluzhna, C. D. Zangmeister, T. C. Allison and Y. Y. J. Tong, J. Am. Chem. Soc., 2012, 134, 1990–1992 Search PubMed.
- M. Di Ventra and N. D. Lang, Phys. Rev. B: Condens. Matter, 2001, 65, 045402 CrossRef.
- T. Weidner, A. Shaporenko, J. Muller, M. Holtig, A. Terfort and M. Zharnikov, J. Phys. Chem. C, 2007, 111, 11627–11635 CrossRef CAS.
- T. Weidner, A. Shaporenko, N. Ballav, A. Ulman and M. Zharnikov, J. Phys. Chem. C, 2008, 112, 1191–1198 Search PubMed.
- T. Weidner, A. Shaporenko, J. Muller, M. Schmid, P. Cyganik, A. Terfort and M. Zharnikov, J. Phys. Chem. C, 2008, 112, 12495–12506 CrossRef CAS.
- T. Nakamura, T. Miyamae, I. Nakai, H. Kondoh, T. Kawamoto, N. Kobayashi, S. Yasuda, D. Yoshimura, T. Ohta, H. Nozoye and M. Matsumoto, Langmuir, 2005, 21, 3344–3353 CrossRef CAS.
- T. Nakamura, T. Miyamae, D. Yoshimura, N. Kobayashi, H. Nozoye and M. Matsumoto, Langmuir, 2005, 21, 5026–5033 CrossRef CAS.
- M. Brust, N. Stuhr-Hansen, K. Nurgaard, J. B. Christensen, L. K. Nielsen and T. Bjornholm, Nano Lett., 2001, 1, 189–191 CrossRef CAS.
- C. K. Yee, A. Ulman, J. D. Ruiz, A. Parikh, H. White and M. Rafailovich, Langmuir, 2003, 19, 9450–9458 CrossRef CAS.
- Y. Negishi, W. Kurashige and U. Kamimura, Langmuir, 2011, 27, 12289–12292 CrossRef CAS.
- S. V. Ley, Comprehensive Organic Functional Group Transformations 2. Synthesis: Carbon with One Heteroatom Attached by a Single Bond, Elsevier Science Ltd. , New York, 1995 Search PubMed.
- J. W. Ciszek, M. P. Stewart and J. M. Tour, J. Am. Chem. Soc., 2004, 126, 13172–13173 CrossRef.
- J. W. Ciszek and J. M. Tour, Chem. Mater., 2005, 17, 5684–5690 CrossRef CAS.
- Y. Choi, Y. Jeong, H. Chung, E. Ito, M. Hara and J. Noh, Langmuir, 2007, 24, 91–96 Search PubMed.
- L. Dreesen, C. Volcke, Y. Sartenaer, A. Peremans, P. A. Thiry, C. Humbert, J. Grugier and J. Marchand-Brynaert, Surf. Sci., 2006, 600, 4052–4057 CrossRef CAS.
- J. K. Lim and S.-W. Joo, Appl. Surf. Sci., 2007, 253, 4830–4835 CrossRef CAS.
- A. Krief, C. Delmotte and W. Dumont, Tetrahedron, 1997, 53, 12147–12158 CrossRef CAS.
- M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shafigullin, I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101, 3706–3712 CrossRef CAS.
- G. C. Lica, B. S. Zelakiewicz and Y. Y. Tong, J. Electroanal. Chem., 2003, 554–555, 127–132 CrossRef CAS.
- M. J. Hostetler, J. J. Stokes and R. W. Murray, Langmuir : the ACS journal of surfaces and colloids, 1996, 12, 3604–3612 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental details and data: Fig. S1–S11. See DOI: 10.1039/c2ra20729j/ |
| This journal is © The Royal Society of Chemistry 2012 |