Novel polyvinyl alcohol based Cr(III)–Sn(IV) doped In(III) nitrate composite foam: synthesis, unit cell formulation and structure†
Nilanjana
Das
,
Sriparna
Chakraborty
and
Prasanta
Kumar Biswas
*
Sol–Gel Division, CSIR-Central Glass & Ceramic Research Institute, 196 Raja S.C. Mullick Road, Jadavpur, Kolkata -700 032, India. E-mail: pkbiswas@cgcri.res.in.; Fax: +91 33 24730957; Tel: +91 33 23223303
First published on 2nd August 2012
Abstract
Nanoscale ordered structures of semiconductors are highly demanding in today's research world for their special optical, electrical and magnetic properties. As a general trend, the inorganic moiety is encapsulated in the PVA host. Encapsulation of the hydrated nitrate of Cr(III), In(III) and Sn(IV) in the crystalline PVA matrix leads to a novel ultra-light, porous and well-structured composite foam material at 100 °C. This is an entirely new composite foam material of inorganic polymer of Sn(IV) and Cr(III) doped In(III) compound in the polymeric chain of PVA. The photoluminescence spectrum confirms atactic-PVA. The structural stability of PVA in the composite foam has been determined by the degree of hydrolysis and the viscosity average molecular weight. FTIR spectroscopy, UV-VIS absorption, elemental and XRD analyses elucidate the unit cell formulation and structure of the composite foam. The microstructural characterizations (SEM, FESEM and HRTEM) explain the porous and branching morphological features.
Introduction
The fabrication of homogeneous and nanoscale ordered structured materials with a specific shape and morphology is a major challenge in today's research world1,2 due to the intake of ordered nanostructured materials in the technological development of the relevant area. Semiconductor particles of the above profile may exhibit unique properties which may be useful in the areas of optoelectronic devices and sensors.3 Indium tin oxide (ITO), being a transparent conducting oxide (TCO), is remarkably known for its transparency and conducting properties.4 It is an n-type material with a body centred cubic structure (a = 10.12 A°)5 and it possesses extensive commercial applications as a component of flat panel displays,6 anti-reflective coatings,7 radiation protection,8 energy efficient windows,9 solar cells,10 memory devices11 and lithium ion battery materials,12 because it has the characteristics of a wide band gap (3.75 eV) with transmissivity in the visible range, low resistivity, strong absorption at ultra-violet wavelengths, high reflectivity in the heat wavelength region and strong attenuation in the microwave region.13–16 Incorporation of magnetic ions like Cr(III), Mn(II), Fe(III) and Ni(II) into the indium oxide matrix has shown ferromagnetism at room temperature, which may be used as a potential material in the domain of dilute magnetic semiconductors (DMS).17 Therefore, selection of this type of semiconductor in the above type of innovation is worthwhile.Utilisation of organic polymers by the soft chemistry route is a prominent method for the synthesis of semiconductor nanoparticles18,19 of intrinsic shapes.20,21 They have been successfully used as the basic matrices22,23 to protect the nanoparticles from undesirable aggregation.24 The use of polyvinyl alcohol (PVA) in this type of synthesis scheme is well known.25 PVA meets all requirements for an ideal temporary technological binder to act as a basic chain-like polymer matrix to host inorganic polymer-like entities with the help of hydrogen-bonding. At the decomposition stage, PVA may be completely removed, leaving behind a nanostructured ordered microstructure of a metal oxide bridged compound. Moreover, this is non-toxic and chemically inert; hence it can be used safely for the ordered structure synthesis. As an example, PVA is used as a template in the preparation of semiconductor nanoparticles of specific shape26,27 and it can also be employed as capping28 as well as pore forming agents.19
Here we have chosen the simple inorganic salts of Cr(III), In(III) and Sn(IV) as starting materials to be incorporated into crystalline PVA in a suitable form. This encapsulation yields a novel material, Cr(III)–Sn(IV) doped hydrated indium(III) nitrate composite foam in a cross-linked PVA matrix (host). This material may have the large possibility of generating an ordered microstructure upon thermal treatment. The unit cell formulation and structure of the inorganic moiety in the PVA crystal is also unexplored. Therefore, in this work, we are the first to report the synthesis, unit cell formulation and structure of the PVA based Cr(III)–Sn(IV) doped In(III) nitrate composite foam.
Experimental section
Synthesis
The hydrated indium nitrate solution was synthesized starting with indium (In) metal ingots (99.99%, SRL, India) and conc. nitric acid (GR grade, E. Merck, India). The initially required amount of indium nitrate solution was taken for the preparation of the precursor. The mixed aqueous–organic based precursor sol (6 wt% equivalent metal oxides, In![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn:Cr = 84.6:9.4:6, at%) was prepared by adding hydrated stannic chloride (SnCl4·5H2O, 98%, Loba Chemie), hydrated chromium nitrate (Cr(NO3)3·9H2O, 99%, ACROS ORGANICS) and the required quantity of water to the above prepared hydrated indium nitrate solution. The solution was evaporated to form a pasty mass, to which a suitable amount of polyvinylalcohol (PVA) (molecular weight 22000, BDH, UK) was added as a temporary organic binder. The solution was concentrated by storing it undisturbed under ambient temperature and atmospheric conditions, when after several days a highly viscous mass was obtained. On complete drying, this composite was heated to 100 °C at a slow heating rate (∼2 °C min−1) to generate the novel PVA encapsulated voluminous Cr(III)–Sn(IV) doped In(III) nitrate composite foam.
:Sn:Cr = 84.6:9.4:6, at%) was prepared by adding hydrated stannic chloride (SnCl4·5H2O, 98%, Loba Chemie), hydrated chromium nitrate (Cr(NO3)3·9H2O, 99%, ACROS ORGANICS) and the required quantity of water to the above prepared hydrated indium nitrate solution. The solution was evaporated to form a pasty mass, to which a suitable amount of polyvinylalcohol (PVA) (molecular weight 22000, BDH, UK) was added as a temporary organic binder. The solution was concentrated by storing it undisturbed under ambient temperature and atmospheric conditions, when after several days a highly viscous mass was obtained. On complete drying, this composite was heated to 100 °C at a slow heating rate (∼2 °C min−1) to generate the novel PVA encapsulated voluminous Cr(III)–Sn(IV) doped In(III) nitrate composite foam.
Characterization
The crystalline phase was identified by X-ray diffractogram (XRD) obtained from an X-ray diffractometer (Philips PW 1730 X-ray diffraction unit employed with a nickel-filtered Cu-Kα radiation source (1.5418 Å radiation)). The diffraction angle was chosen to be in the range 10–70° with continuous scanning at a rate of 0.6° min−1. The morphological features were studied by scanning electron microscopy (SEM) (LEO 400C), field emission scanning electron microscopy (FESEM) (ZEISS, SUPRA™35VP) and transmission electron microscopy (TEM) (TecnaiG2 30.S-Twin, FEI Company, Netherlands). A carbon coated 300 mesh Cu grid was used for TEM images. The vacuum level for this experiment was around 10−9 Torr. For sample preparation, the material was dispersed in cyclohexane, followed by sonication for about 2 h. Finally, it was carefully placed on the Cu-grid. The excess liquid was allowed to evaporate in air. The grids with the sample were examined with an ultra-high resolution (UHR) pole-piece using a LaB6 filament. The operated accelerating voltage and the camera length were 300 kV and 55 cm respectively. The photoluminescence (PL) spectra were recorded using a Perkin Elmer fluorimeter (LS55). The wavelength accuracy of LS55 is ±1.0 nm and its sensitivity is a signal to noise ratio of 500:1 r.m.s. using the Raman band of water with excitation at 350 nm; the excitation and emission band pass is 10 nm. The source for LS55 is a Xenon discharge lamp, equivalent to 20 kW for 8 μs duration. Pulse width at half height is <10 μs. Excitation width and power density were 10 nm and 775 V, respectively, in each case. The elemental analysis of carbon, hydrogen and nitrogen was performed by a 2400 series II CHNS Analyser, Perkin Elmer, USA. Indium, tin and chromium analyses were carried out by the ICP method, iCAP-6800 DWO, Thermofisher Scientific; the uncertainty in each case was 10 ± 2%. FTIR vibrations were measured by IR spectrometer, NICOLET 5700, Thermo Electron Corporation. The number of scans for each experiment was 164 when the wavenumber resolution was 4 cm−1. The absorption spectra were obtained by diffused reflectance measurement by a UV-VIS-NIR spectrophotometer (UV 3600, Shimadzu make, Japan) with ISR 3600 attachment. The apparent viscosities (η) were measured with the help of a Haake make Rheo Stress 6000 viscometer, Thermo Scientific, USA. The temperature was maintained within 25 ± 2 °C. From the apparent viscosity, the relative (ηr) and specific (ηsp) viscosities of pure PVA and the precursor composite were determined by the relations (1) and (2) respectively. | ηr = η/η0 | (1) |
| ηsp = ηr − 1 | (2) |
These two have been used to find out the intrinsic viscosity ([η]int) using Huggin's equation (eqn (3)), which is essential for the determination of the viscosity average molecular weight (Mv) of the system using the Mark–Houwink equation (eqn (4)).
| [η]int = limc→0ηsp/c | (3) |
| [η]int = kMva | (4) |
To understand the hydrolysis reaction of PVA in the presence of the inorganic moiety, the degree of hydrolysis (DH) of pure PVA and the PVA based composite were determined by the equation (eqn (5)).
| DH = 100 − [7.84Sdb/(100 − 0.075Sdb)] | (5) |
The zeta potentials were determined by a zeta potential measurement system, Nano Z, Malvern, UK. Double distilled water was used in the preparation and all the experiments (wherever required). The reproducibility of each and every result was checked by repeating the experiments 2–3 times. Finally, the structure is proposed based on the Avogadro software (Version 1.0.0).
Results and discussion
In the present work, PVA has been chosen as the organic polymer binder, which may act as the host matrix. The hydrated indium(III) nitrate was chosen as the starting material for In2O3, in which hydrated Sn(IV) chloride and Cr(III) nitrate were used as dopants. The above materials were dissolved in double distilled water, which was slowly evaporated followed by heating the solid mass to ∼100 °C. This yielded a novel light-weight, voluminous, highly porous foam-like material (Fig. 1) forming a casing of cross-linked atactic PVA in which Sn(IV) and Cr(III) doped In(III) nitrate are existing through the hydrogen bonding and peroxo linkages. | ||
| Fig. 1 Physical appearance of the novel PVA based Sn(IV) and Cr(III) doped In(III) nitrate composite foam. | ||
The presence of the PVA moiety in the composite foam is evidenced29 by the appearance of a hump at ∼350 nm in the UV-VIS absorption spectrum (measured by the diffused reflectance method, Fig. 2). We worked first to find out the crystalline feature of PVA along with the illustration of the unit cell structure of the polymeric chain. According to Mooney,30 PVA possesses a pseudo-orthorhombic structure, but Bunn31 proposed that it possesses monoclinic structure, which was confirmed by Colvin.32 According to them, strong intermolecular interaction among PVA chains occurs due to intermolecular hydrogen bonding,31 which results in crystalline behavior. But they did not predict the stereoregularity. Later, Assender et al.33 confirmed the stereoregularity in PVA chains. In the present work, incorporation of an inorganic moiety in the PVA host would disrupt the hydrogen bonding in PVA, which may not be effective for complexation with a metal ion. However, re-formation of hydrogen bonding with the inorganic species cannot be ruled out. In our case, the crystallinity of PVA deteriorated in the composite foam, which disrupts hydrogen bonding.34,35 However, the very weak X-ray reflections (Fig. 3b) of the composite foam product observed at 2θ values 15.72°, 18.34° and 21.56° accorded with the work of Colvin32 and Assender et al.33 These weak lines corresponds to the reflections from the planes 〈022〉, 〈202〉 and 〈012〉 of monoclinic PVA, respectively. To make the peaks more prominent (shown by lines i, ii and iii), the Gaussian-fitted result of the XRD spectra (2θ values from 15 to 22) is highlighted in the inset of Fig. 3b. These peaks also confirm the atactic unit cell of PVA.33 In addition, the X-ray reflections at 2θ values 30.50° and 50.79° are in agreement with the cubic bixbyite structure of indium oxide according to JCPDS (card no. 06-0416) for the hexa co-ordinated In(III) complex species in cubic crystal symmetry.
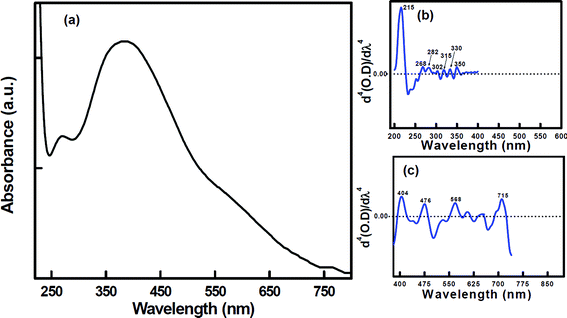 | ||
| Fig. 2 (a) UV-VIS absorption spectrum of the PVA based composite foam material. (b) Fourth derivative of the UV region (200 nm to 400 nm) of (a). (c) Fourth derivative of the visible region (400 nm to 750 nm) of (a). | ||
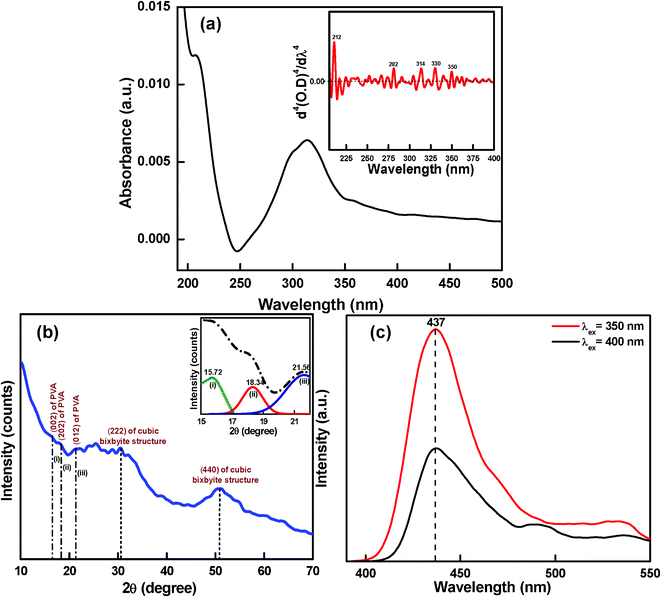 | ||
| Fig. 3 (a) UV-VIS absorption spectrum of the starting PVA material; inset shows the fourth derivative of the spectrum. (b) XRD reflections; inset shows the Gaussian-fitted result of the XRD spectrum (2θ values from 15 to 22). (c) Photoluminescence (PL) spectra of the organic–inorganic composite material. | ||
We have proposed a unit cell structure with the formulation shown in Scheme 1. A series of characterizations such as UV-VIS absorption (Fig. 2), photoluminescence (PL) (Fig. 3c), FTIR spectroscopy (Fig. 4) and elemental analysis (Table 1) have been employed for the elucidation of the unit cell structure (Fig. 5). A unit of the structure may have the values of x = 1, y = 4 and z = 1/4, as formulated from the elemental analysis data (Table 1). The part “x” in the formulation is the probable arrangement of the PVA chain (Fig. 5). The stereoregularity of the –OH groups in PVA generates three types of polymer configuration, which are isotactic, syndiotactic and atactic.36,37 The study of photoluminescence (PL) is a good technique to determine the spatial conformation of PVA.38 We have taken the PL (Fig. 3c) of our composite foam material, which predicts the atactic conformation of PVA, because of the appearance of the emission peak at 437 nm in the PL spectra (excitation wavelengths 350 nm and 400 nm). The existence of cross-linking in the PVA chains was evident from the appearance of the υ(–C–O–C–) vibration39,40 at 1070 cm−1 in the FTIR spectrum (Fig. 4).
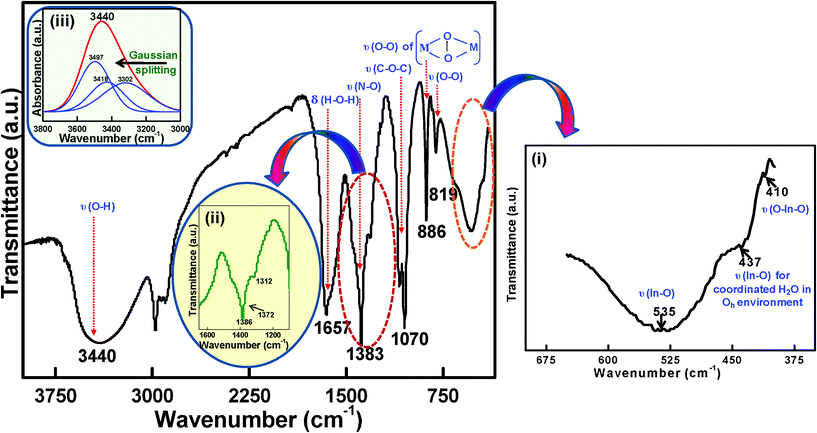 | ||
| Fig. 4 FTIR spectrum of the inorganic moiety incorporated into the PVA matrix; inset (i) highlights the expanded portion of ∼650 to 400 cm−1, inset (ii) highlights the expanded portion of ∼1600 to 1200 cm−1, inset (iii) highlights the Gaussian fitted results from 3800 cm−1 to 3000 cm−1 (absorption mode). | ||
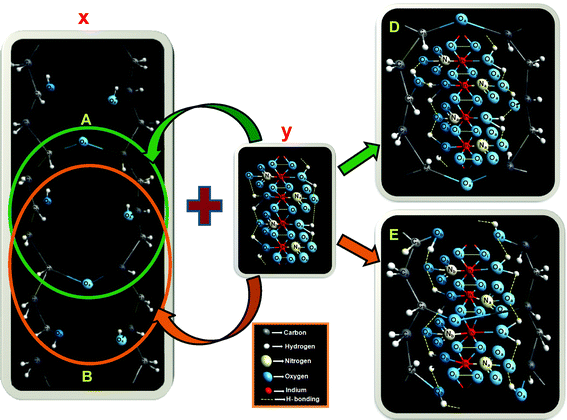 | ||
| Fig. 5 Proposed unit cell structure of the inorganic moiety embedded PVA matrix; “x” shows the probable arrangement of the host PVA chain, “y” shows the inorganic part containing Cr(III)–Sn(IV) doped hydrated indium(III) nitrate, “D” and “E” describe the possible arrangements of “y” embedded in “x”; “O”, “C”, “N” and “In” signify the atoms oxygen, carbon, nitrogen and indium, respectively; “a”, “i” and “b” in the subscripts of the atoms denote the stereochemistry of the atoms as “above the plane”, “in plane” and “below the plane”, respectively. | ||
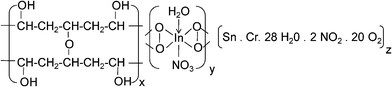 | ||
| Scheme 1 Proposed chemical formulation of the inorganic moiety embedded PVA matrix. Here, x = 1, y = 4 and z = 1/4. | ||
| Elements | Determined from proposed structure (Fig. 5) | Experimental data (uncertainty 10 ± 2%) |
|---|---|---|
| Indium (In) | 32.49 | 32.73 |
| Tin (Sn) | 1.85 | 1.83 |
| Chromium (Cr) | 0.92 | 0.92 |
| Carbon (C) | 6.79 | 6.73 |
| Nitrogen (N) | 4.46 | 4.40 |
| Hydrogen (H) | 2.57 | 2.60 |
To understand the role of hydroxyl groups of PVA in the PVA based composite foam, we have recorded the UV-VIS absorption spectra of the starting PVA material and compared them with those of the composite foam. The absorption spectrum of the dilute aqueous solution (∼10−3 M) of the starting PVA material is shown in Fig. 3a. The spectrum shows two peaks at ∼210 nm and ∼315 nm. To understand the precise peak positions, the fourth derivative41 of the spectrum is shown (inset of Fig. 3a). Here we observe five peaks in the UV-region at 212 nm, 282 nm, 314 nm, 330 nm and 350 nm. In the present case, the n→π* transition at 212 nm in the aqueous solution of PVA is blue shifted if compared with the same transition of pure PVA in dioxane.42 This is possibly due to the solvent effect. Usually hydrogen bonding in the aqueous solution will result in blue shift of the absorption band.43 The bands at 282 nm and 330 nm may be assigned as the π→π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or CC present at the edges of the PVA chain42 in the UV-VIS absorption spectra of the starting PVA material. As we observe five similar peaks in the fourth derivative of the UV-VIS absorption spectra (measured by diffused reflectance method) of the composite foam specimen (Fig. 2b), it would imply that the hydroxyl groups in the composite PVA are hydrogen bonded and do not take part in any bonding with the metal ion. Practically, the spectral pattern exhibits eleven peaks altogether in the fourth derivative of the UV-VIS absorption spectra (measured by the diffused reflectance method) (Fig. 2b and 2c).
O or CC present at the edges of the PVA chain42 in the UV-VIS absorption spectra of the starting PVA material. As we observe five similar peaks in the fourth derivative of the UV-VIS absorption spectra (measured by diffused reflectance method) of the composite foam specimen (Fig. 2b), it would imply that the hydroxyl groups in the composite PVA are hydrogen bonded and do not take part in any bonding with the metal ion. Practically, the spectral pattern exhibits eleven peaks altogether in the fourth derivative of the UV-VIS absorption spectra (measured by the diffused reflectance method) (Fig. 2b and 2c).
To confirm the structural stability of PVA in the composite foam, the degree of hydrolysis (DH) and the viscosity average molecular weight (Mv) were determined. To compare the evaluated values with those of the starting PVA material, the same parameters were determined in the pure system. Detailed characterization techniques are illustrated in the experimental section. The degree of hydrolysis (DH) of the starting PVA material was determined by estimation of the saponification value and loss on drying (LOD) (eqn (5)). Determination of LOD was particularly required to calculate the saponification value to the dried basis (Sdb). The DH of PVA in the composite foam material was also determined following the same procedure. The DH of the starting PVA material was estimated to be ∼98%, whereas in the composite foam the DH decreases to ∼70%, which implies the non-involvement of the metal ions with PVA. The viscosity average molecular weight (Mv) was calculated using the Mark–Houwink equation from the determination of intrinsic viscosity ([η]int, eqn (4)). The values of the Mark–Houwink constants, “k” and “a” for the PVA–water system are assumed to be 2.0 × 10−4 dL g−1 and 0.76, respectively, according to the work of Ambravaneswaran et al.44 The calculated Mv of the starting PVA material and that of the composite foam were found to be around 16500 Dalton and 51000 Dalton respectively.
Based on the above analyses, the decrease in degree of hydrolysis and increase in molecular weight of PVA in composite foam species may be due to the decrease in crystallinity of PVA, because a decrease in crystallinity reduces the degree of hydrolysis.45 There may be some ambiguity about the adsorption of the metal ion above the surface of PVA, as it may also hinder the crystallinity of pure PVA. When PVA gets adsorbed on the surface of the metal ions, the electrokinetic properties of the metal particles change. This can be determined by the study of the zeta-potential46 of the metal system in the presence and in the absence of PVA. In the present case, the zeta-potentials in both cases remain almost equal, which implies the non-involvement of PVA as an adsorbing species onto the metal ion surface.
Once the host's structure is determined, the next target is to find out the possible unit cell structure of the chain-like inorganic network of the complexed species of In(III) to be incorporated in an organized way in the PVA matrix. The inorganic unit cell moiety, “y” is assumed to be sandwiched in between the PVA chains linked by hydrogen bonding, similar to the work of Bunn.31 A single unit may contain four hexa-coordinated In(III) atoms in an octahedral environment,47 which is supported by the appearance of a FTIR vibrational peak at 437 cm−1 (part (i) of Fig. 4), due to the In–O vibration of coordinated water. The In–O bonding nature is also proved by the appearance of υ(In–O) at 535 cm−1 and υ(O–In–O) at 410 cm−148 in the FTIR spectra. Each of the In(III) atoms in the unit cell are linked with one another by peroxo linkages in a side-on manner.49 The existence of the metal–peroxo linkages is proved by the appearance of a FTIR vibration of υ(O–O) at 886 cm−1.50 This can also be proved by the chemical reaction (eqn (6)) between KMnO4 and the material in an acidic medium, where the decolourisation of the 0.01 N KMnO4 solution occurs.
| 2MnO4− + 5O22− + 16H+ → 2Mn2+ + 5O2 + 8H2O | (6) |
The presence of the free NO3− anion (D3h symmetry) is verified by the FTIR peak of υ(N–O) at 1386 cm−151 (Fig. 4). The inset (ii) in Fig. 4 highlights an expanded portion of ∼1600 to 1200 cm−1, where the vibrational frequency at 1372 cm−1 is possibly due to the bonding between the nitrate ion and In(III). This frequency also suggests the formation of hydrogen bonding between the nitrate ion and the water molecule.52 This phenomenon is further supported by the UV-VIS spectrum (Fig. 2). The fourth derivative41 of the spectrum of the composite foam in the UV region shows an additional peak at 302 nm, which may be due to the presence of the hydrogen bonded nitrate ion (NO3−), as the peak due to pure NO3− generally appears at 313 nm.53,54 The blue shifting of the peak position confirms the presence of a hydrogen bonded nitrate ion (NO3−).
To understand the different vibrational modes of water, the FTIR spectrum was measured with the absorption mode, and the Gaussian fitted results were determined from 3800 cm−1 to 3000 cm−1 (inset (iii) of Fig. 4). The vibrational peaks at 3497 cm−1, 3412 cm−1 and 3302 cm−1 conclude the presence of water molecules co-ordinated with the metal atoms and also the hydrogen bonding behavior of the hydrogens of water molecule (possible hydrogen bonds are shown by dotted lines in Fig. 5). The presence of physically adsorbed water molecules cannot be ruled out as a number of stretching vibrations due to υ(O–H) appears.
Sn(IV) and Cr(III) will occupy the positions of In(III) after an interval of about four “y” units. However, a question may arise regarding the driving force of the replacement of In(III) by Sn(IV) and Cr(III). The redox potentials of the couples In(III)/In, Sn(IV)/Sn(II) and Cr(III)/Cr are −0.34V, +0.15V and −0.74V, respectively. The redox potential of the In(III)/In couple has to be lowest in order to be replaced by Sn(IV) and Cr(III). This may be possible if In(III) undergoes any complexation, which may reduce the formal potential of In(III)/In even below −0.74V in the solution phase. Reduction of the formal potential of the In(III)/In couple to −0.768 V in the presence of the complex [InCl5]2− has been reported by Yang et al.55 In the solution phase, the possibility of the existence of several complex species (eqn (7)–(10)) cannot be ruled out:56–58
| 3In3+ + 4H2O → In3(OH)45+ + 4H+ | (7) |
| In3+ + Cl− → InCl2+ | (8) |
| InCl2+ + H2O → InClOH+ + H+ | (9) |
| InCl2+ + In3+ + H2O → In2ClOH4+ + H+ | (10) |
The presence of these complex species may reduce the formal potential of the In(III)/In couple to the desired level to allow its replacement by Sn(IV) and Cr(III). In addition, as the ionic radius of Cr(III) (∼75.5 pm) is smaller than that of In(III) (∼94 pm), interstitial replacement of In(III) may be feasible, provided the charge remains the same. According to the report of Lincoln,59 the water exchange rate constant for In(III) (∼104 s−1) is much higher than that of Cr(III) (∼10−6 s−1). Cr(III), being a d3 system, forms a very stable complex with water due to the large ligand field stabilization energy. All these factors may help in the replacement of In(III) by Cr(III) in the inorganic chain structure. However, in the case of doping with Sn(IV), the mechanism will be somewhat different as there is a difference in charge, although the ionic radius of Sn(IV) (∼83 pm) is close to that of In(III) (∼94 pm). The mechanism of In(III) replacement by Sn(IV) is predicted in Scheme 2. Here the lone pairs of the coordinated water molecule may be utilized to stabilize the excess positive charge generated by the substitution of Sn(IV), thereby reducing H2O to OH−. This may be justified from the work of Alshehri et al.,60 where there is evidence for the existence of [Sn(OH)6]2− in the solid state.61 However, for thin films, the surface Sn atoms are not likely to attract and activate H2O to form OH, as shown by Anderson et al.62
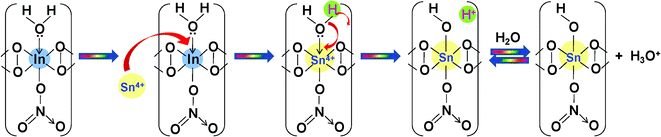 | ||
| Scheme 2 Proposed mechanism of In(III) replacement by Sn(IV). | ||
Among the seven peaks in the UV region of the composite foam, six have already been assigned; the residual peak ∼270 nm may be due to the presence of the hexa-coordinated polymeric Sn–O–Sn type moiety.63 The fourth derivative of the absorption peaks at 404 nm, 476 nm, 568 nm and 715 nm in the visible region highlights the d–d transitions of Cr(III) in the octahedral environment.64–66 The co-ordination of water and NO3− ions with the metal ions (already discussed) may be further supported by the co-ordination behavior of the incorporated Cr(III), with the help of the fourth derivative of the electronic spectra (visible region). Cr(III), being a transition metal, exhibits d–d transitions, which have become an important tool to understand the nature of bonding existing here in the system. Cr3+ (d3 system) generally exhibits three spin-allowed transitions, 4A2g (F)→4T2g (F), 4A2g (F)→4T1g (F) and 4A2g (F)→4T1g (P).67 Among these, the first two transitions generally fall in the visible region. When Cr(III) becomes co-ordinated with water, the transitions generally appear at ∼575 nm and ∼400 nm, respectively.68 Hence, the appearance of peaks at 404 nm and 568 nm may be due to the d–d transitions of Cr(III) as a result of co-ordinated water. On the other hand, NO3− is a weaker field ligand than water.69 As a result, the transitions are expected to be red shifted when Cr(III) is bonded with NO3−. Therefore, the peaks at 476 nm and 715 nm may be due to the d–d transitions of Cr(III) as a result of linkage with the NO3− ion. Therefore, we can infer that Cr(III) is co-ordinated with water molecules and a NO3− ion. Thus, Cr(III) incorporation helped us to elucidate the basic structure of co-ordinated In(III) and Sn(IV), as we may presume that doping of Cr(III) only replaces In(III) from its lattice position, without any distortion of its basic molecular structure. Although the doping quantity of Cr(III) is much lower than In(III) and Sn(IV), it is sufficient to understand the structural features. In(III) and Sn(IV) being non-transition metals, this phenomenon of d–d transitions is not possible.
Now, the unit containing the inorganic entity “y” may be present inside the PVA network in two possible arrangements. In one arrangement, “y” might be incorporated in between two cross-linking (–C–O–C–) moieties, resulting in the structure “D” (shown in Fig. 5), while in another arrangement “C” might be incorporated in such a way that the cross-linking (–C–O–C–) remains at the centre of the inorganic moiety, resulting in the structure “E”. Both the predicted structures have the same structural formulation, but it is very difficult to say which one of them exists in reality. The elemental compositions determined from the probable structures are comparable with the experimental results of chemical analysis (Table 1), where the error for each element is within 2%. We have also tried to highlight the stereochemistry of the probable structure. “a”, “i” and “b” in the suffixes of the atoms, as shown in Fig. 5, denote the stereoregularity of the atoms as “above the plane”, “in plane” and “below the plane”, respectively.
The part “z” shown in the formulation is not incorporated in the structure. The FTIR band at 3415 cm−1 (inset (iii) of Fig. 4), 1312 cm−1 (inset (ii) of Fig. 4) and 819 cm−1 (Fig. 4) may be due to the presence of physically adsorbed water molecules,70 free NO271 and free υ(O–O) vibration of the trapped oxygen atoms,72 respectively, which are shown as “z” in the formulation.
The microstructural characterizations explain very well the above morphological features of the above described material, which is physically manifested as branched fibers. The porous nature can clearly be seen in the SEM image (Fig. 6a and 6b). Fig. 6a shows large pores with a fiber-like morphology. Fig. 6b is the magnified image of Fig. 6a, where the pores and the fibrous nature are more clearly visible. This fiber-like feature is distinctly visible in the FESEM image of the specimen (Fig. 6c). We also observe branching in the fibers. We may assume that these fibers are composed of repeating units of our proposed structure, as established in Fig. 5. The TEM image (Fig. 6d) shows the formation of small particles. The inset of the figure highlights the histogram of the nano-clusters of average size ∼3 nm, which are aligned, implying formation of the fiber-like material. The SAED pattern (Fig. 6e) determined from the TEM image shows the diffraction indices, which are in agreement with those of the planes determined from the XRD diffractogram (Fig. 3b). The HRTEM image (Fig. 6f) reflects the 〈202〉 plane of crystalline PVA, which confirms the existence of PVA as proposed by Colvin, where a very strong reflection was obtained from the 〈202〉 plane assigned as the 2θ line at 19.26°, equivalent to the d-spacing 4.6 Å.
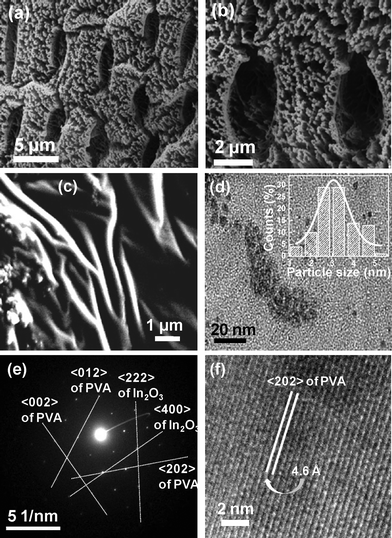 | ||
| Fig. 6 (a) SEM image highlighting large pores with fiber-like morphology. (b) Magnified image of (a) where the pores and the fibrous nature are more clearly visible. (c) FESEM image of the specimen highlighting branching in the fibers. (d) TEM image showing the formation of small particles, inset highlights the histogram of the nano-clusters of average size ∼3 nm. (e) SAED pattern determined from the TEM image highlighting the diffraction indices. (f) HRTEM image reflecting the 〈202〉 plane of crystalline PVA. | ||
The present process that will be involved in the formation of an ordered porous semiconductor from this novel organic–inorganic composite foam material is beautifully explained with the help of Scheme 3. (A) represents the unit cell structure as shown in Fig. 5. Now a number of (A) moieties may be arranged as shown in (B). Again there might be a large number of such entities present. Upon subjecting the organic–inorganic composite material to a certain temperature (say, 400 °C), the organic part will be decomposed, resulting in the formation of a porous aggregated inorganic part (C). Interestingly, when this composite foam material was cured at 400 °C, a similar type of nanoparticles aggregated with porous morphology was obtained, (D). However, detailed study of (D) and the sequential steps of oxide transformation from this novel foam-type precursor and evaluation of their structure is under progress, which will be reported in a forthcoming article.
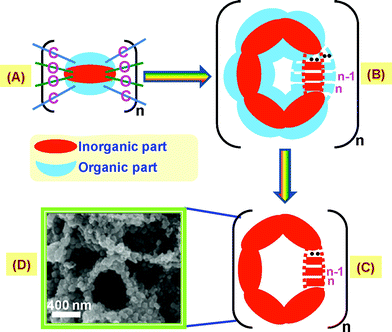 | ||
| Scheme 3 Proposed process that may be involved in the formation of an ordered porous semiconductor from this foam-type voluminous inorganic moiety embedded PVA matrix. | ||
Conclusion
In conclusion, a novel PVA encapsulated inorganic moiety (Cr(III)–Sn(IV) doped In(III) nitrate) is reported, along with its unit cell formulation and structure utilizing photoluminescence spectra, FTIR and UV-VIS absorption spectroscopy. The unit cell formulation obtained from the probable structure was verified from the elemental analysis. This new foam-like material is a lightweight and highly porous substance, where PVA is not involved with the metal ion. The morphology of the specimen is highlighted by the SEM, FESEM and TEM images. The features of crystalline PVA could be noted in this foam specimen, as evident from the XRD diffractogram, SAED and HRTEM images.Acknowledgements
The authors wish to acknowledge the Director, CGCRI for permission to publish this work. One of the authors (ND) thanks Board of Research in Nuclear Science (BRNS), Govt. of India and Council of scientific and Industrial Research (CSIR) for offering her SRF fellowship.References
- S. W. Kim, M. Kim, W. Y. Lee and T. Hyeon, J. Am. Chem. Soc., 2002, 124, 7642–7643 CrossRef CAS.
- X. Gao, Y. Cui, R. M. Levenson, L. W. K. Chung and S. Nie, Nat. Biotechnol., 2004, 22, 969–976 CrossRef CAS.
- N. Pinna, G. Neri, M. Antonietti and M. Niederberger, Angew. Chem., Int. Ed., 2004, 43, 4345–4349 CrossRef CAS.
- S. Luo, K. Okada, S. Kohik, F. Tsutsui, H. Shimooka and F. Shoji, Mater. Lett., 2009, 63, 641–643 Search PubMed.
- L. Dai, X. L. Chen, J. K. Jian, M. He, T. Zhou and B. Q. Hu, Appl. Phys. A: Mater. Sci. Process., 2002, 75, 687–689 CrossRef CAS.
- M. Sawada, M. Higuchi, S. Kondo and H. Saka, Jpn. J. Appl. Phys., 2001, 40, 3332–3336 Search PubMed.
- H. Kim, J. S. Horwitz, W. H. Kim, Z. H. Kafafi and D. B. Chrisey, J. Appl. Phys., 2002, 91, 5371–5376 CrossRef CAS.
- R. A. Synowicki, J. S. Hale, N. J. Ianno and J. A. Woollam, Surf. Coat. Technol., 1993, 62, 499–503 Search PubMed.
- I. Hambergend and C. G. Granquist, J. Appl. Phys., 1996, 60, R123–160 Search PubMed.
- P. Stulik and J. Singh, Sol. Energy Mater. Sol. Cells, 1996, 40, 239–251 Search PubMed.
- K. S. Yook, J. L. Lee, S. H. Kim and J. Jang, Appl. Phys. Lett., 2008, 92, 223305–223307 Search PubMed.
- D. W. Kim, I. S. Hwang, S. J. Kwon, H. Y. Kang, K. S. Park, Y. J. Choi, K. J. Choi and J. G. Park, Nano Lett., 2007, 7, 3041–3045 CrossRef CAS.
- X. U. Bao-qiang, F. Rui-kang, Y. Bin and D. Yong, Trans. Nonferrous Met. Soc. China, 2010, 20, 643–648 Search PubMed.
- B. Zhou, R. Q. Cui, Q. J. Pang, Y. D. Wang, F. Y. Meng, T. T. Sun, Z. M. Ding and X. B. Yu, Appl. Surf. Sci., 2001, 172, 245–252 Search PubMed.
- P. K. Biswas, A. De, K. Ortner and S. Korder, Mater. Lett., 2004, 58, 1540–1545 Search PubMed.
- S. S. Kim, Y. C. Yoon and K. H. Kim, J. Electroceram., 2003, 10, 95–101 Search PubMed.
- S. Kundu, D. Bhattacharya, J. Ghosh, P. Das and P. K. Biswas, Chem. Phys. Lett., 2009, 469, 313–317 Search PubMed.
- P. Reiss, E. Couderc, J. D. Girolamo and A. Pron, Nanoscale, 2011, 3, 446–489 RSC.
- I. Truijen, M. K. V. Bael, H. V. Rul, J. D. Haen and J. Mullens, J. Sol-Gel Sci. Technol., 2007, 43, 291–297 Search PubMed.
- X. Li, T. Duan, X. Zhu and Y. Qian, Mater. Lett., 2006, 60, 3350–3353 Search PubMed.
- H. G. Zhang, Q. Zhu and Y. Wang, Chem. Mater., 2005, 17, 5824–5830 CrossRef CAS.
- Y. Zhou, S. Yu, C. Wang, X. Li, Y. Zhu and Z. A. Chen, Chem. Commun., 1999, 1229 RSC.
- W. Mahler, Inorg. Chem., 1988, 27, 435–436 CrossRef CAS.
- T. Teranishi, I. Kiyokawa and M. Miyake, Adv. Mater., 1998, 10, 596–599 CrossRef CAS.
- N. T. Andrianov, S. R. A. Gavad and N. V. Zinkova, Steklo Keram., 2006, 12, 20–23 Search PubMed.
- S. W. Choi, J. Y. Park, C. Lee, J. G. Lee and S. S. Kim, J. Am. Ceram. Soc., 2011, 94, 1974–1977 CrossRef CAS.
- J. D. Bass, C. D. Schaper, C. T. Rettner, N. Arellano, F. H. Alharbi, R. D. Miller and H. C. Kim, ACS Nano, 2011, 5, 4065–4072 Search PubMed.
- T. K. Kundu, N. Karak, P. Barik and S. Saha, IJSCE., 2011, 1, 19–24 Search PubMed.
- A. M. Shehap, Egypt. J. Solids., 2008, 31, 75–91 Search PubMed.
- R. C. L. Mooney, J. Am. Chem. Soc., 1941, 63, 2828–2832 Search PubMed.
- C. W. Bunn, Nature, 1948, 161, 929–930 CrossRef CAS.
- B. G. Colvin, Nature, 1974, 248, 756–759 Search PubMed.
- H. E. Assender and A. H. Windle, Polymer, 1998, 39, 4303–4312 Search PubMed.
- X. D. Ma, X. F. Qian, J. Yin, H. A. Xi and Z. K. Zhu, J. Colloid Interface Sci., 2002, 252, 77–81 CrossRef CAS.
- X. F. Qian, J. Yin, Y. F. Yang, Q. H. Lu, Z. K. Zhu and J. Lu, J. Appl. Polym. Sci., 2001, 82, 2744–2749 CrossRef CAS.
- J. H. Choi, S. W. Ko, B. C. Kim, J. Blackwell and W. S. Lyoo, Macromolecules, 1999, 34, 2964–2972 Search PubMed.
- P. D. Hong, C. M. Chou and W. T. Chuang, J. Appl. Polym. Sci., 2001, 79, 1113–1120 Search PubMed.
- S. Ram and T. K. Mandal, Chem. Phys., 2004, 303, 121–128 Search PubMed.
- J. Gong, L. Luo, S. H. Yu, H. Qianab and L. Fei, J. Mater. Chem., 2006, 16, 101–105 RSC.
- H. S. Mansur, R. L. Orifice and A. P. Mansur Alexander, Polymer, 2004, 45, 7193–7202 CrossRef CAS.
- Y. L. Yeow, S. Azali, S. Y. Ow, M. C.L. Wong and Y. K. Leong, Talanta, 2005, 68, 156–164 Search PubMed.
- E. Rusu, A. Airinei, V. Barboui and D. Timpu, J. Optoelectron. Adv. M., 2007, 9, 1044–1047 Search PubMed.
- S. Singh, A. S. N. Murthy and C. N. R. Rao, Trans. Faraday Soc., 1966, 62, 1056–1066 RSC.
- B. Ambravaneswaran, E. Wilkes and O. Basaran, Phys. Fluids, 2002, 14, 2606–2621 Search PubMed.
- B. Briscoe, P. Luckham and S. Zhu, Polymer, 2000, 41, 3851–3860 CrossRef CAS.
- M. Wiśniewska, Colloid Polym. Sci., 2011, 289, 341–344 Search PubMed.
- J. Mink, C. Nemeth, L. Hajba, M. Sandstrom and P. L. Goggin, J. Mol. Struct., 2003, 661–662, 141–151 CrossRef CAS.
- T. Sato, J. Therm. Anal. Calorim., 2005, 82, 775–782 Search PubMed.
- V. Mahadevan, M. J. Henson, E. I. Solomon and T. D. P. Stack, J. Am. Chem. Soc., 2000, 122, 10249–10250 CrossRef CAS.
- M. Saleem, M. Sharma, H. N. Sheikh and B. L. Kalsotra, Indian J. Chem., 2007, 46A, 1423–1426 Search PubMed.
- W. W. Rudolph, D. Fischer, M. R. Tomneyc and C. C. Pyec, Phys. Chem. Chem. Phys., 2004, 6, 5145–5155 RSC.
- D. J. Goebbert, E. Garand, T. Wende, R. Bergmann, G. Meijer, K. R. Asmis and D. M. Neumark, J. Phys. Chem. A, 2009, 113, 7584–7592 CrossRef CAS.
- C. N. R. Rao, Ultra-violet and visible spectroscopy-Chemical applications, Butterworth & Co. (Publishers) Ltd, London, 3rd edn, 1975 Search PubMed.
- H. J. McConnell, J. Chem. Phys., 1952, 20, 700 CAS.
- M. H. Yang and I. W. Sun, J. Chinese Chem. Soc., 2004, 51, 253–260 Search PubMed.
- G. Biedermann, Studies on the Hydrolysis of Metal Ions. Part 14. The Hydrolysis of the Indium(III) Ion, In3, Arkiv For Kemi, 1956, 9, 277–292 Search PubMed.
- D. Ferri, Acta Chem. Scand., 1972a, 26, 733–746 Search PubMed.
- D. Ferri, Acta Chem. Scand., 1972b, 26, 747–759 Search PubMed.
- S. F. Lincoln, Helv. Chim. Acta, 2005, 88, 523–545 CrossRef CAS.
- S. Alshehri, J. Burgess, J. Fawcett, S. A. Parsons and D. R. Russell, Polyhedron, 2000, 19, 399–405 Search PubMed.
- R. Scholder and G. Brauer, Handbook of Preparative Inorganic Chemistry, Academic Press, New York, 2nd edn, 1965, vol. 2, pp. 1677 Search PubMed.
- A. B. Anderson, S. Seong and E. Grantscharova, J. Phys. Chem., 1996, 100, 17535–17538 Search PubMed.
- Z. C. Liu, H. R. Chen, W. M. Huang, J. L. Gu, W. B. Bu, Z. L. Hua and J. L. Shi, Microporous Mesoporous Mater., 2006, 89, 270–275 Search PubMed.
- V. Thomas, G. Jose, P. L. Paulose and N. V. Unnikrishnan, Mater. Chem. Phys., 2002, 77, 826–830 Search PubMed.
- S. R. Ramanan and D. Ganguli, J. Non-Cryst. Solids, 1997, 212, 299–302 Search PubMed.
- G. Calas, O. Majerus, L. Galoisy and L. Cormier, Chem. Geol., 2006, 229, 218–226 Search PubMed.
- V. Singh, R. P. S. Chakradhar, J. L. Rao and J. J. Zhu, Mater. Chem. Phys., 2008, 111, 143–148 Search PubMed.
- G. Dong, X. Xiao, M. Peng, Z. Ma, S. Ye, D. Chen, H. Qin, G. Deng, Q. Liang and J. Qiu, RSC Adv., 2012, 2, 2773–2782 RSC.
- R. R. Crichton, Biological Inorganic Chemistry: An Introduction, Elsevier, 2008, 22 Search PubMed.
- W. Di, X. Wang, G. Pan, X. Bai, B. Chen and X. Ren, Chem. Phys. Lett., 2007, 436, 129–132 Search PubMed.
- P. S. Ajitha and M. K. M. Nair, RJPBCS., 2010, 1, 449–459 Search PubMed.
- Y. C. Hong and H. S. Uhm, Scr. Mater., 2008, 59, 262–264 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra20916k/ |
| This journal is © The Royal Society of Chemistry 2012 |