Combination of organic cation and cyclic sulfonylamide anion exhibiting plastic crystalline behavior in a wide temperature range†
Makoto
Moriya
*ab,
Takaaki
Watanabe
a,
Wataru
Sakamoto
a and
Toshinobu
Yogo
*a
aDivision of Nanomaterial Science, EcoTopia Science Institute, Nagoya University, 464-8603, Furo-cho, Chikusa-ku, Nagoya, Japan. E-mail: moriya@esi.nagoya-u.ac.jp; yogo@esi.nagoya-u.ac.jp; Fax: 81 52 789 2121; Tel: 81 52 789 2750
bJapan Science and Technology Agency, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama, Japan 332-0012
First published on 25th July 2012
Abstract
Organic ionic plastic crystals (OIPCs) with cyclic perfluoroalkyl sulfonylamide as a counter anion were synthesized with an intention of expanding the temperature range of their plastic crystalline phase. The thermal analysis of the obtained OIPCs showed a solid–solid phase transition below room temperature and a high melting point above 250 °C, that indicated their plastic crystalline behavior in a wide temperature range, as well as thermal durability up to approximately 370 °C. The electrochemical measurements of these OIPCs revealed the solid-state ionic conductivity of the plastic crystalline phase and a wide electrochemical window between 0.4 and 6.2 V vs. Li/Li+.
Introduction
Flexible solid materials with high thermal stability and ionic conductivity are considered as candidates of novel electrolytes to achieve next-generation batteries with high reliability and large energy density.1 Plastic crystals (PCs) consisting of organic anions and cations, which are called organic ionic plastic crystals (OIPCs), have thus attracted considerable attention.2 Since a plastic crystalline phase that shows disorder in molecular orientation or conformation within the crystalline lattice is a mesophase between liquid and crystal, these OIPCs show not only a long-range ordered structure like a crystal but also the dynamic behavior of the component building blocks.3 Because of these characteristics, the synthesis of novel OIPCs and their application in lithium ion batteries are actively studied.4–27 Some OIPCs containing lithium salts show high ionic conductivity in the solid state at ambient temperature.4,23–27 Furthermore, OIPCs exhibit mechanical flexibility that reduces the interfacial resistance between electrolyte and electrodes. These features are ideal for employing OIPCs as matrix materials in solid electrolytes.These OIPCs generally exhibit high pyrolysis temperatures above 300 °C but most of them have a relatively low melting point around 100 °C.4–16,24–27 These characteristics make them insufficiently safe for batteries and other devices such as fuel cells and solar cells. Therefore, a strategy to increase the usable temperature range of plastic crystalline materials as solid electrolytes is necessary. However, controlling the physical properties of OIPCs is still difficult because of the lack of material design guidelines. Indeed, only a few OIPCs have been synthesized to date exhibiting plastic crystalline behavior under ambient temperature with melting points greater than 200 °C.17–23 As almost all OIPCs exhibiting such high melting points possess anions with a symmetrical structure, for example, PF6−, BF4−, Cl−, Br− or I−, the strategy to increase the melting point of OIPCs is limited.17,19–23 In order to obtain OIPCs with high ionic conductivity, bis(trifluoromethanesulfonyl)amide anion, N(SO2CF3)2− (TFSA−), has been widely used because of its structural flexibility and high dissociativity derived from the sulfonylamide moiety with electron-withdrawing groups.4–6 Owing to the steric and electronic features of TFSA−, OIPCs with TFSA− often exhibit a low-temperature phase transition from crystal to PC. However, this feature causes a decrease in the melting point. In order to increase the usable temperature range of PCs, it is necessary to increase the melting point and lower the temperature of the crystal to PC phase transition.
Hence, we attempted to use N(SO2CF2)2CF2− (CPFSA−), which is an analogue of TFSA−, as a counter anion, to develop novel PCs with high melting points and low-temperature phase transitions (Fig. 1). CPFSA− has a cyclic structure combining sulfonylamide groups and an electron-withdrawing perfluoroalkyl chain. Our strategy for the molecular design of OIPCs focuses on the steric and electronic structure of the anion to elevate the melting point of OIPCs. The restricted structural flexibility of the CPFSA anion because of its cyclic structure is used to increase the melting point of OIPCs. The large dissociativity of the anion derived from the sulfonylamide group with the electron-withdrawing moiety is employed to exhibit the low-temperature phase transition from crystal to plastic crystal. We report the synthesis of novel OIPCs by using CPFSA− and quaternary alkylammonium cations. The thermal behavior and ionic conductive properties of the obtained OIPCs are also described. We found the obtained OIPCs to exhibit uniform and consistent plastic crystalline behavior in a wide temperature range and solid-state ionic conductivity.
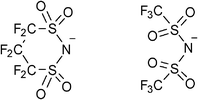 | ||
| Fig. 1 Molecular structures of N(SO2CF2)2CF2− (CPFSA−) (left) and N(SO2CF3)2− (TFSA−) (right). | ||
Experimental
Lithium 1,1,2,2,3,3-hexafluoropropane-1,3-disulfonamide (LiCPFSA), lithium bis(trifluoromethanesulfonyl)amide (LiTFSA), tetramethylammonium bromide, ethyltrimethylammonium iodide, diethylmethylamine, triethylmethylammonium chloride, tetraethylammonium bromide, methyl iodide and DMSO-d6 were purchased from Tokyo Chemical Industry Co. Lithium foil for the electrochemical measurements was purchased from Honjo Chemical Co.1H and 19F NMR spectra were recorded with a JEOL JNM-A400. 13C NMR spectra were recorded with a JEOL JNM-GSX270. 1H and 13C NMR spectra were referenced to the natural-abundance proton or carbon signal of the solvent employed. 19F NMR spectra were referenced to C6H5CF3 as an external standard. X-Ray diffraction patterns were measured using Rigaku SmartLab diffractometer using Cu-Kα radiation with a monochromator. DSC analyses were performed using a SII EXSTAR DSC6220 with a heating rate of 10 °C min−1 in N2 with Al2O3 as a reference. TG–DTA analyses were performed with a Rigaku Thermo plus EVO with a heating rate of 10 °C min−1 in O2 using Al2O3 as a reference. Elemental analyses were performed on a Perkin Elmer 2400II. Ionic conductivities were measured using disks of electrolyte sandwiched between two SUS plates in a two-electrode cell and sealed in a closed vessel. The vessel was placed into a bench-top type temperature chamber (ESPEC, SU-241). Conductivity data were collected by ac impedance measurements carried out with a frequency response analyzer (Biologic, VMP3). The electrochemical stability was evaluated by linear sweep voltammetry using a three-electrode cell. The disk of the obtained OIPC was sandwiched between a SUS disc, used as the working electrode, and lithium foil, used as the counter and reference electrodes. The measurement was carried out on an electrochemical interface (Biologic, VMP3) with a scan rate of 0.1 mV s−1. All cells for the electrochemical experiments were assembled in an argon-filled glovebox (UNIlab2000, MBraun) and equilibrated at the operating temperature for at least 3 h before taking any measurements.
[N1122][I]
[N1122][I] was prepared according to the reported procedure.28 1.0 mL (16.1 mmol) of methyl iodide was added dropwise into an acetonitrile solution of 1.59 mL (13.4 mmol) of diethylmethylamine. The mixture was stirred at room temperature for 24 h. Then the solvent was removed under reduced pressure. The obtained solid was washed with 5 mL of hexane at least six times to remove any hexane-soluble impurities. The product was dried under vacuum at 100 °C for 48 h. The product yield was 65.6%.N,N-Dimethylpyrrolidinium iodide and N,N-ethylmethyl pyrrolidinium iodide were prepared using the same procedure by substituting the appropriate primary amine.
[N1111][CPFSA]
0.491 g of tetramethylammonium bromide (3.19 mmol) was dissolved in 2 mL of distilled water. 0.951 g of LiCPFSA was dissolved in 6 mL of distilled water. The two aqueous solutions were mixed and then stirred at room temperature for 3 h to produce a white precipitate. The precipitate was collected by filtration and washed twice with distilled water to remove any water-soluble impurities. The product was dried under vacuum at 100 °C for more than 48 h. The product yield was 72%. 1H NMR (400 MHz, rt, DMSO-d6): δ 3.08 (s, 12H, NCH3). 13C NMR (68 MHz, rt, DMSO-d6): δ 54.5 (t, JCN = 3.9 Hz, NCH3), 109.3 (tt, 1JCF = 272.9 Hz, 2JCF = 25.4 Hz, –CF2CF2CF2–), 112.4 (tt, 1JCF = 296.6 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–). 19F NMR (376 MHz, rt, DMSO-d6): δ −120.4 (br s), −105.4 (br s). 19F NMR (565 MHz, rt, CD3OD): δ −120.8 (br s), −127.3 (br s). 19F NMR (565 MHz, −80 °C, CD3OD): δ −116.0 (d, JFF = 242.7 Hz, –CF2CF2CF2–), −116.5 (d, JFF = 231.4 Hz, –CF2CF2CF2–), −127.0 (d, JFF = 259.7 Hz, –CF2CF2CF2–), −138.4 (d, JFF = 282.3 Hz, –CF2CF2CF2–). Anal. Calc. for C7H12F6N2O4S2: C, 22.95; H, 3.30; N, 7.65. Found: C, 23.12; H, 3.20; N, 7.46%.In all of the following, the same procedure as above for [N1111][CPFSA] was used, substituting the appropriate quaternary ammonium halide starting material.
[N1112][CPFSA]
1H NMR (400 MHz, rt, DMSO-d6): δ 1.23 (t, 3H, JHH = 7.3 Hz, NCH2CH3), 3.00 (s, 9H, NCH3), 3.31 (q, 2H, JHH = 7.3 Hz, NCH2CH3). 13C NMR (67.8 MHz, rt, DMSO-d6): δ 7.9 (s, NCH2CH3), 51.7 (t, JCN = 4.2 Hz, NCH3), 61.0 (t, JCN = 2.8 Hz, NCH2CH3), 109.4 (tt, 1JCF = 272.9 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–), 112.5 (tt, 1JCF = 297.7 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–). 19F NMR (376 MHz, rt, DMSO-d6): δ −120.4 (br), −105.4 (br). Anal. Calc. for C8H14F6N2O4S2: C, 25.26; H, 3.71; N, 7.37. Found: C, 25.27; H, 3.87; N, 7.56%.[N1122][CPFSA]
1H NMR (400 MHz, rt, DMSO-d6): δ 1.22 (t, 6H, JHH = 6.8 Hz, NCH2CH3), 2.94 (s, 6H, NCH3), 3.29 (q, 4H, JHH = 6.8 Hz, NCH2CH3). 13C NMR (67.8 MHz, rt, DMSO-d6): δ 7.6 (s, NCH2CH3), 48.9 (t, JCN = 4.2 Hz, NCH3), 58.0 (t, JCN = 3.1 Hz, NCH2CH3), 109.4 (tt, 1JCF = 272.9 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–), 112.5 (tt, 1JCF = 297.7 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–). 19F (376 MHz, rt, DMSO-d6): δ −120.5 (br), −105.5 (br). Anal. Calc. for C9H16F6N2O4S2: C, 27.41; H, 4.09; N, 7.10. Found: C, 27.34; H, 4.02; N, 7.16%.[N1222][CPFSA]
1H NMR (400 MHz, rt, DMSO-d6): δ 1.19 (t, 9H, JHH = 7.3 Hz, NCH2CH3), 2.86 (s, 3H, NCH3), 3.24 (q, 6H, JHH = 7.3 Hz, NCH2CH3)3). 13C NMR (67.8 MHz, rt, DMSO-d6): δ 7.2 (s, NCH2CH3), 45.9 (t, JCN = 4.2 Hz, NCH3), 55.1 (t, JCN = 2.8 Hz, NCH2CH3), 109.5 (tt, 1JCF = 272.7 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–), 112.5 (tt, 1JCF = 298.0 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–). 19F NMR (376 MHz, rt, DMSO-d6): δ −120.4 (br), −105.4 (br). Anal. Calc. for C10H18F6N2O4S2: C, 29.41; H, 4.44; N, 6.86. Found: C, 29.40; H, 4.36; N, 6.74%.[N2222][CPFSA]
1H NMR (400 MHz, rt, DMSO-d6): δ 1.17 (t, 12H, JHH = 6.8 Hz, NCH2CH3), 3.21 (q, 8H, JHH = 6.8 Hz, NCH2CH3). 13C NMR (68 MHz, rt, DMSO-d6): δ 6.9 (s, NCH2CH3), 51.5 (t, JCN = 3.1 Hz, NCH2CH3), 109.3 (tt, 1JCF = 272.9 Hz, 2JCF = 25.4 Hz, –CF2CF2CF2–), 112.4 (tt, 1JCF = 296.6 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–). 19F NMR (376 MHz, rt, DMSO-d6): δ −120.4 (br), −105.5 (br). Anal. Calc. for C11H20F6N2O4S2: C, 31.28; H, 4.77; N, 6.63. Found: C, 31.30; H, 4.74; N, 6.70%.[Pyr11][CPFSA]
1H NMR (400 MHz, rt, DMSO-d6): δ 2.10 (m, 4H, N(–CH2CH2CH2CH2–)), 3.08 (s, 6H, NCH3), 3.44 (m, 4H, N(–CH2CH2CH2CH2–)). 13C NMR (68 MHz, rt, DMSO-d6): δ 21.4 (s, N(–CH2CH2CH2CH2–)), 51.1 (t, JCN = 3.9 Hz, NCH3), 64.9 (t, JCN = 3.1 Hz, N(–CH2CH2CH2CH2–)), 109.3 (tt, 1JCF = 272.6 Hz, 2JCF = 25.7 Hz, –CF2CF2CF2–), 112.5 (tt, 1JCF = 298.0 Hz, 2JCF = 25.1 Hz, –CF2CF2CF2–). 19F NMR (376 MHz, rt, DMSO-d6): δ −120.4 (br), −105.3 (br). Anal. Calc. for C9H14F6N2O4S2: C, 27.55; H, 3.60; N, 7.14. Found: C, 27.56; H, 3.46; N, 7.11%.[Pyr12][CPFSA]
1H NMR (400 MHz, rt, DMSO-d6): δ 1.24–1.27 (m, 3H, NCH2CH3), 2.05 (m, 4H, N(–CH2CH2CH2CH2–)), 2.94 (s, 3H, NCH3), 3.32–3.42 (m, 6H, N(–CH2CH2CH2CH2–) and NCH2CH3). 13C NMR (68 MHz, rt, DMSO-d6): δ 8.7 (s, NCH2CH3), 21.1 (s, N(–CH2CH2CH2CH2–)), 47.0 (t, JCN = 3.9 Hz, NCH3), 58.5 (t, JCN = 3.1 Hz, NCH2CH3), 62.0 (t, JCN = 3.3 Hz, N(–CH2CH2CH2CH2–)), 109.3 (tt, 1JCF = 272.6 Hz, 2JCF = 25.7 Hz, –CF2CF2CF2–), 112.4 (tt, 1JCF = 297.6 Hz, 2JCF = 25.7 Hz, –CF2CF2CF2–). 19F NMR (376 MHz, rt, DMSO-d6): δ −120.5 (br), −105.5 (br). Anal. Calc. for C10H16F6N2O4S2: C, 29.56; H, 3.97; N, 6.89. Found: C, 29.52; H, 3.82; N, 6.81%.Results and discussion
Synthesis and characterization
In this study, we selected alkylammonium or pyrrolidinium cation to synthesize OIPCs with a wide potential window. These OIPCs were obtained through the anion metathesis reaction between ammonium or pyrrolidinium halide and LiCPFSA (Scheme 1). The reaction proceeded at room temperature in aqueous solution to give a white solid product. The obtained OIPCs were collected by filtration and dried under reduced pressure at 100 °C for more than 48 h to remove water.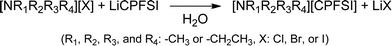 | ||
| Scheme 1 Syntheses of organic ionic plastic crystals consisting of ammonium cation and CPFSA anion. | ||
The 13C NMR spectrum of the tetramethylammonium salt [N1111][CPFSA] yielded three signals attributable to the methyl carbon of the ammonium cation at 54.5 ppm and two peaks assignable to the CPFSA anion at 109.3 and 112.4 ppm. In the 19F NMR of this compound two broad peaks were observed at −120.4 and −105.4 ppm. In the 19F NMR spectrum measured at −80 °C, the shape and chemical shift of these signals changed to four doublets at −116.0, −116.5, −127.0 and −138.4 ppm with an intensity ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2:1 as a result of vicinal fluorine–fluorine coupling (Fig. S1. ESI†). The peaks at −116.0 and −127.0 ppm are attributable to the CF2 group in the center of the ring, and the signals at −116.5 and −138.4 ppm are assignable to the CF2 groups adjacent to the sulfonyl group. These results support the formation of the target compounds. Furthermore, the NMR spectrum indicates that the CPFSA anion shows the molecular motion in the solution state similar to the conformational change between chair and boat forms.
:1:2:1 as a result of vicinal fluorine–fluorine coupling (Fig. S1. ESI†). The peaks at −116.0 and −127.0 ppm are attributable to the CF2 group in the center of the ring, and the signals at −116.5 and −138.4 ppm are assignable to the CF2 groups adjacent to the sulfonyl group. These results support the formation of the target compounds. Furthermore, the NMR spectrum indicates that the CPFSA anion shows the molecular motion in the solution state similar to the conformational change between chair and boat forms.
The formation of ethyltrimethyl-, diethyldimethyl-, triethylmethyl- and tetraethyl-ammonium salts, which are abbreviated as [N1112][CPFSA], [N1122][CPFSA], [N1222][CPFSA] and [N2222][CPFSA], respectively, were also confirmed from the NMR measurements and elemental analysis. We also synthesized dimethylpyrrolidinium salts, [Pyr11][CPFSA], and ethylmethylpyrrolidinium salts, [Pyr12][CPFSA], using a similar procedure.
In the powder X-ray diffraction analysis, several sharp and strong reflections were observed for these ion combinations at room temperature (Fig. S2, ESI†). This result shows that these compounds possess long-range ordered structure in the solid state. Hence, the obtained ion combinations do not exist as amorphous or supercooled states, but exist as crystals or plastic crystals at ambient temperature.
Thermal behavior
The thermal durability of these ammonium salts was evaluated by means of thermogravimetry analysis (TGA) under oxygen flow. The results indicated that the obtained OIPCs with CPFSA− are thermally stable up to approximately 370 °C. As depicted in Fig. 2, [N1222][CPFSA] shows comparable thermal stability compared with that of the corresponding TFSA salts.![TG curves of [N1222][CPFSA] (—) and [N1222][TFSA] ().](/image/article/2012/RA/c2ra20945d/c2ra20945d-f2.gif) | ||
Fig. 2 TG curves of [N1222][CPFSA] (—) and [N1222][TFSA] (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ). ). | ||
The DSC curves of [N1222][CPFSA], [N1122][CPFSA] and [N1112][CPFSA] measured from −100 to 330 or 380 °C under nitrogen flow are shown in Fig. 3. The obtained OIPCs showed two endothermic peaks in the measured temperature range. For example, [N1222][CPFSA] afforded peaks at −28.2 and 268.3 °C. We confirmed that the first peak represents the solid–solid phase transition and the second peak corresponds to melting. [N1122][CPFSA] and [N1112][CPFSA] also showed similar thermal behavior. The peaks for the solid–solid phase transition and melting of [N1122][CPFSA] were observed at −21.4 and 307.2 °C and those of [N1112][CPFSA] at 7.2 and 361.5 °C, respectively. Small entropy values of 7.4, 9.1 and 5.4 J K−1 mol−1 were evaluated from the second peaks of [N1222][CPFSA], [N1122][CPFSA] and [N1112][CPFSA], respectively. This result suggests the existence of a plastic crystalline phase in these OIPCs since these values are less than the criterion set by Timmermans29: ΔSm < 20 J K−1 mol−1. Interestingly, the obtained OIPCs with CPFSA− show uniform and consistent plastic crystalline behavior in a wide temperature range without any phase transitions, which is a rare thermal behavior for OIPCs.9 From a device perspective, this characteristic is important and highly useful.
![DSC curves of [N1222][CPFSA], [N1122][CPFSA] and [N1112][CPFSA].](/image/article/2012/RA/c2ra20945d/c2ra20945d-f3.gif) | ||
| Fig. 3 DSC curves of [N1222][CPFSA], [N1122][CPFSA] and [N1112][CPFSA]. | ||
With the decrease in the number of ethyl groups at the nitrogen center, both the temperature of solid–solid phase transition and the melting point of [N1112][CPFSA], [N1122][CPFSA] and [N1222][CPFSA] increase. This indicates that the molecular dynamics of the ethyl groups in the crystal lattice are critical to the physical properties of CPFSA salts. The ammonium salts with CPFSA− show much higher melting points than the salts consisting of the corresponding ammonium cation and TFSA−. The melting points of [N1222][TFSA], [N1122][TFSA] and [N1112][TFSA] are reported to be 96, 98 and 109 °C, respectively.5,6 The melting point of [N1112][CPFSA] exceeded that of [N1112][TFSA] by more than 250 °C. The thermal behavior of these OIPCs revealed by the TG and DSC measurements suggests that the use of CPFSA− instead of TFSA− increases the melting point, while retaining the low-temperature phase transition point from the crystal to PC without loss of thermal durability. Ammonium salts with homoleptic cations, [N1111][CPFSA] and [N2222][CPFSA], did not show melting behavior in the measured temperature range (Fig. S3, ESI†).
The pyrrolidinium salts, [Pyr11][CPFSA] and [Pyr12][CPFSA], also show thermal behavior similar to that of the ammonium CPFSA salts (Fig. 4). Low-temperature phase transitions at 20.8 and −15.8 °C were observed for [Pyr11][CPFSA] and [Pyr12][CPFSA], respectively. The melting point of [Pyr12][CPFSA] was observed at 302.4 °C. The entropy value for the melting of [Pyr12][CPFSA] was calculated to be 12.4 J mol−1 K−1, which was less than the above-mentioned criterion of Timmermans. Similar to the thermal behavior of [N1111][CPFSA] and [N2222][CPFSA], the melting point of [Pyr11][CPFSA] was not observed between −100 and 380 °C.
![DSC curves of [Pyr11][CPFSA] and [Pyr12][CPFSA].](/image/article/2012/RA/c2ra20945d/c2ra20945d-f4.gif) | ||
| Fig. 4 DSC curves of [Pyr11][CPFSA] and [Pyr12][CPFSA]. | ||
The obtained OIPCs show plastic crystalline behavior in a considerably wide temperature range. This distinguishing thermal behavior is derived from the steric and electrostatic feature of CPFSA−. The existence of the sulfonylamide group causes the large ionic dissociativity to lower the phase transition temperature. The cyclic structure of this anion suppresses the molecular dynamics of the anionic moiety and increases the melting point. It is expected that the six-membered ring of the CPFSA anion is fixed in a chair-form structure without conformational change to a boat-form structure in a plastic crystalline phase. This structural feature of CPFSA− results in completely different thermal behavior compared with that of OIPCs with I−, BF4−, SCN−, N(CN)2− and TFSA−. In the DSC curves of [Pyr11][X] (X = I−, BF4−, SCN−, N(CN)2− and TFSA−), multiple solid–solid phase transitions were observed indicating the complex molecular dynamics of the pyrrolidinium cation.30 In contrast, the obtained OIPCs with the CPFSA anion show a single solid–solid phase transition behavior before melting. This suggests that the molecular dynamics of the cation units in the obtained compounds are constrained from those of OIPCs with the above-mentioned anions, I−, BF4−, SCN−, N(CN)2− and TFSA− because of the bulkiness of the CPFSA anion.
Ionic conductivity and electrochemical window
The ionic conductivity of the OIPCs with the CPFSA anion was measured by using a disk, which was produced by pressing the powder of the products. The disk was placed between a pair of stainless-steel electrodes in the cell used for the ac impedance measurements. The fabrication of the measurement cell was performed in an argon-filled glove box to avoid contamination by moisture. The ac impedance data showed a well-defined semicircle and a low-frequency spike that indicate the considerably small grain boundary resistance of the electrolyte.The obtained PCs exhibited ionic conductive properties above their solid–solid phase transition temperature (Fig. 5). The conductivity correlates with the observations from the thermal analysis. PCs with low-temperature phase transitions tend to show high ionic conductivities. [N1222][CPFSA], which possesses the lowest phase transition point of the obtained PCs in this study, shows solid-state ionic conductivity at −20 °C. The ionic conductivities continuously increased with increasing temperature since the obtained PCs with the CPFSA anion show single solid–solid phase transition behavior, whereas the corresponding TFSA salts afforded stepwise increase of ionic conductivity associated with multiple phase transition behavior.
![Solid-state ionic conductivities of the synthesized ammonium salts with CPFSA anion: [N1222][CPFSA] (●), [N1122][CPFSA] (○) and [N1112][CPFSA] (■), as a function of temperature.](/image/article/2012/RA/c2ra20945d/c2ra20945d-f5.gif) | ||
| Fig. 5 Solid-state ionic conductivities of the synthesized ammonium salts with CPFSA anion: [N1222][CPFSA] (●), [N1122][CPFSA] (○) and [N1112][CPFSA] (■), as a function of temperature. | ||
[Pyr11][CPFSA] and [Pyr12][CPFSA] also show solid-state ionic conductivity under the plastic crystalline phase (Fig. 6). Similar to those of ammonium CPFSA salts, [Pyr12][CPFSA] with a low-temperature solid–solid phase transition showed higher ionic conductivity than that of [Pyr11][CPFSA]. In general, the anionic species of OIPCs greatly affect the solid-state ionic conductivity. For example, it is reported that [Pyr11][BF4], [Pyr11][SCN] and [Pyr11][N(CN)2] exhibited similar ionic conductivity at ambient temperature, whereas the conductivities of [Pyr11][TFSA] and [Pyr11][I] were lower than those of the above-mentioned three pyrrolidinium salts.30 Compared with the reported value of ionic conductivity for dimethylpyrrolidinium salts, the solid-state ionic conductivity of [Pyr11][CPFSA] was slightly higher than that of [Pyr11][TFSA] and [Pyr11][I] and lower than that of [Pyr11][BF4], [Pyr11][SCN] and [Pyr11][N(CN)2].
![Solid-state ionic conductivities of the synthesized pyrrolidinium salts with CPFSA anion, [Pyr11][CPFSA] (■) and [Pyr12][CPFSA] (●), as a function of temperature.](/image/article/2012/RA/c2ra20945d/c2ra20945d-f6.gif) | ||
| Fig. 6 Solid-state ionic conductivities of the synthesized pyrrolidinium salts with CPFSA anion, [Pyr11][CPFSA] (■) and [Pyr12][CPFSA] (●), as a function of temperature. | ||
Ionic conductivities under high-temperature conditions were measured for [N1222][CPFSA] and [N1122][CPFSA] (Fig. 7 and supplementary information, Fig. S4, ESI†). These compounds showed solid-state ionic conductivity above 200 °C. High ionic conductivities were observed below the melting points of these compounds, which are evaluated to be 8 × 10−5 S cm−1 at 250 °C and 2 × 10−4 S cm−1 at 290 °C for [N1222][CPFSA] and [N1122][CPFSA], respectively. Clearly, it is desirable to employ OIPCs as thermally durable solid electrolytes.
![Solid-state ionic conductivities of [N1222][CPFSA] (●) as a function of temperature from −40 to 250 °C.](/image/article/2012/RA/c2ra20945d/c2ra20945d-f7.gif) | ||
| Fig. 7 Solid-state ionic conductivities of [N1222][CPFSA] (●) as a function of temperature from −40 to 250 °C. | ||
The electrochemical stability of [N1222][CPFSA] is shown in Fig. 8. The measurements were carried out using three-electrode cells with a stainless-steel working electrode and lithium foil as the counter and reference electrodes. The measurements revealed a wide stability window for [N1222][CPFSA] between approximately 0.4 and 6.2 V vs. Li/Li+. This result indicates that OIPCs with the CPFSA anion possess similar electrochemical stability to that of TFSA salts.5
![Linear sweep voltammogram of [N1222][CPFSA] with a scan rate of 0.1 mV s−1 measured at 100 °C.](/image/article/2012/RA/c2ra20945d/c2ra20945d-f8.gif) | ||
| Fig. 8 Linear sweep voltammogram of [N1222][CPFSA] with a scan rate of 0.1 mV s−1 measured at 100 °C. | ||
Conclusions
In this study, we synthesized novel OIPCs consisting of ammonium cations with a CPFSA− with sulfonylamide group and cyclic structure. The substitution of TFSA− by CPFSA− as a counter-anion of OIPCs resulted in a dramatic increase of the melting point and a low-temperature solid–solid phase transition. The obtained OIPCs exhibited plastic crystalline behavior and solid-state ionic conductivity in a wide temperature range above 200 °C. The electrochemical stability of [N1222][CPFSA] was estimated to be between 0.4 and 6.2 V vs. Li/Li+. Our strategy to control the physical properties of OIPCs by structurally controlling the component units, focusing on the steric and electronic character, has enabled the development of novel ionic conductive materials with high thermal durability and mechanical flexibility derived from the plastic crystalline behavior.Acknowledgements
We thank Prof. H. Suzuki's laboratory at Dept. Appl. Chem., Tokyo Inst. Technol. for the elemental analyses. This work was supported by the JST PRESTO program.References
- (a) J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS; (b) M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS.
- (a) D. R. MacFarlane and M. Forsyth, Adv. Mater., 2001, 13, 957 CrossRef CAS; (b) J. M. Pringle, P. C. Howlett, D. R. MacFarlane and M. Forsyth, J. Mater. Chem., 2010, 20, 2056 RSC.
- J. N. Sherwood, Plastically Crystalline State; Orientationally Disordered Crystals, John Wiley & Sons Ltd, London, 1979 Search PubMed.
- D. R. MacFarlane, J. H. Huang and M. Forsyth, Nature, 1999, 402, 792 CrossRef CAS.
- (a) D. R. MacFarlane, P. Meakin, J. Sun, N. Amini and M. Forsyth, J. Phys. Chem. B, 1999, 103, 4164 CrossRef CAS; (b) D. R. MacFarlane, J. Sun, J. Golding, P. Meakin and M. Forsyth, Electrochim. Acta, 2000, 45, 1271 CrossRef; (c) D. R. MacFarlane, P. Meakin, N. Amini and M. Forsyth, J. Phys.: Condens. Matter, 2001, 13, 8257 CrossRef CAS.
- H. Matsumoto, M. Yanagida, K. Tanimoto, M. Nomura, Y. Kitagawa and Y. Miyazaki, Chem. Lett., 2000, 922 CrossRef CAS.
- E. I. Cooper and C. A. Angell, Solid State Ionics, 1986, 18–19, 570 CrossRef CAS.
- K. M. Johansson, J. Adebahr, P. C. Howlett, M. Forsyth and D. R. MacFarlane, Aust. J. Chem., 2007, 60, 57 CrossRef CAS.
- S. A. Forsyth, K. J. Fraser, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Green Chem., 2006, 8, 256 RSC.
- (a) Z. B. Zhou, H. Matsumoto and K. Tatsumi, Chem.–Eur. J., 2006, 12, 2196 CrossRef CAS; (b) T. Enomoto, S. Kanematsu, K. Tsunashima, K. Matsumoto and R. Hagiwara, Phys. Chem. Chem. Phys., 2011, 13, 12536 RSC.
- S. A. Forsyth, S. R. Batten, Q. Dai and D. R. MacFarlane, Aust. J. Chem., 2004, 57, 121 CrossRef CAS.
- S. A. Forsyth and D. R. MacFarlane, J. Mater. Chem., 2003, 13, 2451 RSC.
- P. M. Dean, B. R. Clare, V. Armel, J. M. Pringle, C. M. Forsyth, M. Forsyth and D. R. MacFarlane, Aust. J. Chem., 2009, 62, 334 CrossRef CAS.
- J. Adebehr, M. Grimsley, N. M. Rocher and D. R. MacFarlane, Solid State Ionics, 2008, 178, 1798 CrossRef.
- (a) G. Annat, J. Adebahr, I. R. McKinnon, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2007, 178, 1065 CrossRef CAS; (b) A. J. Seeber, M. Forsyth, C. M. Forsyth, S. A. Forsyth, G. Annat and D. R. MacFarlane, Phys. Chem. Chem. Phys., 2003, 5, 2692 RSC.
- F. U. Shah, S. Glavatskih, P. M. Dean, D. R. MacFarlane, M. Forsyth and O. N. Antzutkin, J. Mater. Chem., 2012, 22, 6928 RSC.
- T. Shimizu, S. Tanaka, N. Onoda-Yamamuro, S. Ishimaru and R. Ikeda, J. Chem. Soc., Faraday Trans., 1997, 93, 321 RSC.
- T. Tanabe, D. Nakamura and R. Ikeda, J. Chem. Soc., Faraday Trans., 1991, 87, 987 RSC.
- M. Tansho, D. Nakamura and R. Ikeda, Ber. Bunsen-Ges. Phys. Chem., 1991, 95, 1643 CrossRef CAS.
- M. Hattori, S. Fukada, D. Nakamura and R. Ikeda, J. Chem. Soc., Faraday Trans., 1990, 86, 3777 RSC.
- (a) L. Jin, P. Howlett, J. Efthimiadis, M. Kar, D. MacFarlane and M. Forsyth, J. Mater. Chem., 2011, 21, 10171 RSC; (b) S. Forsyth, J. Golding, D. R. MacFarlane and M. Forsyth, Electrochim. Acta, 2001, 46, 1753 CrossRef CAS.
- J. Golding, N. Hamid, D. R. MacFarlane, M. Forsyth, C. Forsyth, C. Collins and J. Huang, Chem. Mater., 2001, 13, 558 CrossRef CAS.
- J. Sun, D. R. MacFarlane and M. Forsyth, Solid State Ionics, 2002, 148, 145 CrossRef CAS.
- (a) Y. Abu-Lebdeh, P. J. Alarco and M. Armand, Angew. Chem., Int. Ed., 2003, 42, 4499 CrossRef CAS; (b) P. J. Alarco, Y. Abu-Lebdeh and M. Armand, Solid State Ionics, 2004, 175, 717 CrossRef CAS; (c) Y. Abu-Lebdeh, P. J. Alarco, A. Abouimrane, L. Ionescu-Vasii, A. Hammami and M. Armand, J. New Mater. Electrochem. Syst., 2005, 8, 197 CAS.
- (a) J. Huang, M. Forsyth and D. R. MacFarlane, Solid State Ionics, 2000, 136–137, 447 CrossRef CAS; (b) P. C. Howlett, D. R. MacFarlane and A. F. Hollenkamp, Electrochem. Solid-State Lett., 2004, 7, A97 CrossRef CAS; (c) P. C. Howlett, N. Brack, A. F. Hollenkamp, M. Forsyth and D. R. MacFarlane, J. Electrochem. Soc., 2006, 153, A595 CrossRef CAS; (d) P. C. Howlett, Y. Shekibi, D. R. MacFarlane and M. Forsyth, Adv. Eng. Mater., 2009, 11, 1044 CrossRef CAS; (e) M. Yoshizawa-Fujita, D. R. MacFarlane, P. C. Howlett and M. Forsyth, Electrochem. Commun., 2006, 8, 445 CrossRef CAS.
- T. Rüther, J. Huang and A. F. Hollenkamp, Chem. Commun., 2007, 5226 RSC.
- M. Moriya, D. Kato, W. Sakamoto and T. Yogo, Chem. Commun., 2011, 47, 6311 RSC.
- J. Sun, D. R. MacFarlane and M. Forsyth, Ionics, 1997, 3, 356 CrossRef CAS.
- J. Timmermans, J. Phys. Chem. Solids, 1961, 18, 1 CrossRef CAS.
- J. Adebah, M. Forsyth and D. R. MacFarlane, Electrochim. Acta, 2005, 50, 3853 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 19F NMR spectrum of [N1111][CPFSA] in CD3OD at −80 °C, XRD patterns of [N1111][CPFSA], [N1112][CPFSA], [N1122][CPFSA], [N1222][CPFSA], [N2222][CPFSA], [Pyr11][CPFSA] and [Pyr12][CPFSA] at room temperature, DSC curves of [N1111][CPFSA] and [N2222][CPFSA], solid-state ionic conductivities of [N1122][CPFSA] as a function of temperature from 50 to 290 °C. See DOI: 10.1039/c2ra20945d/ |
| This journal is © The Royal Society of Chemistry 2012 |