Water dissociation on a gold cluster: the effect of carbon nanostructures as a substrate
Naresh K
Jena
ab,
K R S
Chandrakumar
*a and
Swapan K
Ghosh
*ab
aTheoretical Chemistry Section, Chemistry Group, Bhabha Atomic Research Centre, Mumbai-400 085, India. E-mail: krsc@barc.gov.in; Fax: +91 22 25505151; Tel: +91 22 25595092
bHomi Bhabha National Institute, Mumbai, India. E-mail: skghosh@barc.gov.in
First published on 23rd August 2012
Abstract
Water dissociation at the gold interface can be of fundamental importance in areas like catalysis and electrochemistry. In recent years, gold nanocluster based catalysts have opened up new avenues of research. In the present computational study, we have investigated the catalytic activity of a pristine gold cluster Au8 as well as the cluster supported on substrates such as defective graphene and a carbon nanotube (CNT) for the dissociation of a water molecule. Our results reveal that the dissociation of water on the pristine cluster can be associated with higher barrier height while the process becomes more facile with the carbon nanostructures as substrates, especially the CNT. For the Au8–CNT system, we have demonstrated the dissociation of a second water molecule is associated with a lower barrier than the first one and this leads to the generation of molecular hydrogen. We attribute the strong charging effects of the cluster in case of the Au8–CNT system as the possible driving factor for this process. The CNT–Au system is, thus, proposed as a robust nanostructure for the water dissociation process. Although gold cluster based catalysis has been well explored in terms of their favorable role in other reactions, our report comes as a first report towards the demonstration of catalytic activity of nanoscale gold for the water dissociation process.
1. Introduction
The generation of clean and green energy is one of the most sought after areas of research being pursued world-wide very intensely and actively. Depletion of fossil fuels and CO2 induced climate change have spearheaded the need for green energy sources. To this end, hydrogen holds promise as a potential energy carrier. The direct thermal splitting of water, H2O → H2 + 1/2O2 seems promising as it does not generate CO2 but produces hydrogen, however this process is associated with a large kinetic barrier requiring temperatures in excess of 2000 °C.1,2 Similarly, the electrocatalysis of water is energy intensive with a very low yield and also requires expensive catalysts.2 Hence, the onus is on the researchers to design robust and efficient catalysts for water dissociation.The interaction of water with metals is of central importance in areas such as catalysis, electrochemistry and corrosion etc.2,3 In several industrially relevant catalytic processes viz. the water gas shift reaction, steam reforming etc., the formation and dissociation of water is a vital step.2,3 It is, therefore, of fundamental interest to study water’s interactions with metal surfaces which can help us to improve the performance of catalysts relevant to these processes.
In recent years, there have been several theoretical and experimental studies on transition metal based nanostructures for high-capacity hydrogen storage and catalysts for water dissociation.2–7 Phatak et al.4 have discussed the adsorption and dissociation of a water monomer on (111) surfaces of several transition metal atoms such as Cu, Au, Pt, Pd, and Ni. The first principles calculations of Pozzo et al.6 shed light on the dissociation of water on Rh(111) and Ni(111) surfaces. They have calculated the barriers for water dissociation on Rh and Ni surfaces to be 0.92 and 0.89 eV respectively.
In electrochemical reactions, water dissociation at the electrode interfaces to form H and OH which are key intermediates in the oxidation or reduction reactions, is an extremely important step. In fuel cells, the efficiency of the oxygen reduction reaction (ORR) taking place at the electrode (cathode) is greatly influenced by the interfacial water. Water facilitates the oxidation of metal in presence of dissolved O2 which in turn limits the performance of fuel cells as the catalysis of the ORR cells takes place on the metallic surfaces.8,9 The higher oxidation potential of gold makes it a potentially better candidate than Pt which is commonly used in fuel cells. Interestingly, it has been shown that deposition of gold clusters on Pt leads to improved stability of a Pt-ORR electrocatalyst.5
Over the years, the surprising activity of gold for oxidation reactions has come to the forefront.10 The pioneering work of Haruta et al. on the discovery of the high catalytic activity of Au-nanoparticles towards the CO oxidation reaction spearheaded the larger and burgeoning role of nanoscale gold in surface science and catalysis.11 Recently, selective oxidation of alcohols on gold leading to the formation of esters, aldehydes and acids has grabbed much attention.12–14 To get fundamental insights into nanoscale catalysis, gold clusters offer interesting possibilities as model systems.15–17
Date et al. have shown the vital role of H2O on promoting the rate of the CO oxidation reaction on Au nanoparticles.18 These authors attribute the enhancement in reactivity of gold on the basis of formation of OH groups and an activated O atom from the reaction of H2O with O2 on the perimeter of gold nanoparticles. The exact role, however, of H2O in facilitating the CO oxidation reaction on nanoscale gold is still under debate.18–20 Employing all-electron relativistic DFT calculations, Kuang et al. have studied the adsorption of H2O molecules on Aun (n = 1–13) clusters and have reported an enhancement in the reactivity of the water molecule.21 It is, therefore, of fundamental importance to study the dissociation of water on nanoscale gold. The preceding discussions which enunciate the importance of interactions of water with gold prompted us to explore this in greater detail. Hence, following this line of thought, in the current investigation we have tried to address the dissociation process of a single water molecule on a gold cluster Au8. Apart from considering the water dissociation reaction on the bare cluster, we have also considered the water dissociation on this cluster supported on surfaces like graphene and a carbon nanotube (CNT).22–25 These two surfaces representing the two fascinating types of carbon nanostructures lie at the helm of the affairs in modern day nanoscience and technology. From our important observations, we demonstrate that, while the water dissociation on the pristine cluster can have a higher barrier, the latter can be significantly reduced by considering substrates like graphene and CNTs. Particularly, a gold cluster supported on a CNT can be considered as a better nanostructure for water dissociation leading to the generation of hydrogen molecule.
2. Computational details
All the calculations have been performed using the electronic structure software TURBOMOLE.26 We have employed the def2-SVP basis set for the atoms of the substrate (graphene and the CNT) and def2-TZVP for all other atoms. Relativistic corrected effective core potential ecp-mwb has been used for 60 core electrons of the Au atom. The Perdew–Burke–Ernzerhof (PBE) density functional has been used for all the calculations. The model graphene surface consists of 27 carbon atoms with a vacancy site and the peripheral carbon atoms saturated by 14 hydrogen atoms (Fig. 1(a)). Likewise, to model the CNT surface we have considered a small segment of CNT(5,0) with a single vacancy site and the terminal C-atoms saturated by H-atoms (Fig. 1(b)). The potential energy surface for the water dissociation reaction on Au8 adsorbed on these surfaces is computed by considering one of the O–H bonds in a water molecule as the reaction coordinate. The geometry corresponding to maxima on the potential energy surface is taken as the initial guess for the transition state (TS) optimization. The optimized TS is confirmed by a hessian calculation where only one imaginary frequency is observed. The barrier for water dissociation (ΔEa) is calculated by subtracting the energy of the TS and initial state (IS). The final state is designated by FS.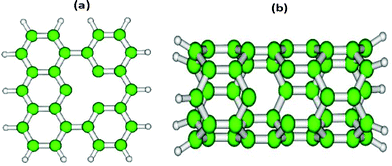 | ||
| Fig. 1 Graphical representations of two model carbon nanostructure surfaces; (a) defective graphene and (b) defective CNT(5,0). | ||
3. Results and discussion
Let us begin our discussion with the consideration of the water dissociation reaction on a gas phase Au8 cluster. The initial geometry for this gold cluster has Td symmetry and serves as an ideal model gold cluster which has exceptionally high stability.27 Taking this geometry of the cluster Au8 as a model one, we have previously explored the catalytic properties with reference to CO oxidation.28 The water molecule can interact with this highly symmetric gold cluster at two types of symmetry unique gold atoms (differing in coordination number) in the cluster. It is found that a single water molecule favorably interacts with the low-coordinated gold atom which is presented in Fig. 2 (IS). The binding of a water molecule with the gold cluster is favorable and the corresponding binding energy is −0.37 eV. In the IS geometry, the Au–O (interacting with O-atom of H2O) bond distance is found to be 2.455 Å. Similarly the O–H distance of the bound water molecule is 0.974 Å and the H–O–H angle is 105.27°. The potential energy surface for the dissociation of the water molecule has been generated by choosing one of the O–H bonds as the reaction coordinate. The optimized TS is presented in Fig. 2. It is noted that in the TS structure, one of the O–H bond distances (the reaction coordinate) is found to be 1.929 Å whereas the other O–H distance remains unaltered (0.978 Å). In the FS geometry, the water molecule is completely dissociated into H and OH which are bound to gold atoms through a bridging mode. The estimated barrier for water dissociation is 2.04 eV. Let us analyze our results more carefully by comparison with some earlier reported results for water dissociation. The dissociation of water on the (111) surfaces of Rh and Ni have been previously examined by DFT methods.6 These metals are important from the viewpoint of their utility as catalysts in several industrially relevant processes. A comparison, therefore, can give us important insights into the performance of these catalysts. The estimations of binding energies of a water molecule on Rh and Ni surfaces are reported to be 0.35 and 0.25 eV respectively.6 Our calculated binding energy (0.37 eV) matches closely with the above results. Thus, it is inferred that water can favorably interact with nanoscale gold and the interaction energies are comparable with that of other metals like Rh and Ni. The barriers for water dissociation on the above two surfaces have been reported to be 0.92 and 0.89 eV respectively for Rh and Ni.6 Our result for the barrier of water dissociation (2.04 eV) is much higher as compared to the above results. We, thus infer that although the water molecule binds to the gold cluster favorably, the barrier to dissociate the water molecule into H and OH is larger as compared to other common metals employed in catalysis.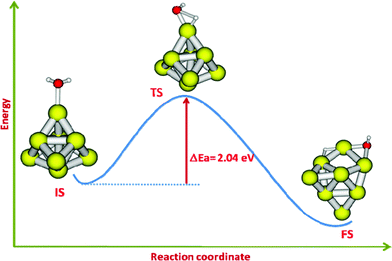 | ||
| Fig. 2 The reaction pathway showing IS, TS and FS for the dissociation of a single water molecule on the Au8 cluster. | ||
To facilitate the chemical reactions on gold nanoclusters, we can take a cue from the favorable role of substrates in promoting their catalytic activity. The higher barrier for the water dissociation process, in principle, can be altered by taking suitable substrates for the gold clusters. We have considered two model carbon nanostructure surfaces, viz. graphene and a CNT which can be promising candidates as substrates. These two surfaces have a single vacancy in the form of defect. It is noted that the interactions of perfect graphene/CNT with an Au cluster are very weak. For better adherence and effective tuning of the electronic environment of the active catalyst, surface vacancies can be a useful concept. Recently Kauffman et al. have explored the sensory applications of Au decorated CNTs.29 They have shown that the binding of a gold cluster at the vacancy site is stronger and the sensing property for gases like CO is also significantly modulated by the presence of a vacancy site on the CNT surface.29 Hence, considering a gold cluster adsorbed on top of a vacancy site on graphene or a CNT can be a good choice of nanostructure for water dissociation. The water dissociation pathways (IS, TS and FS) on the defective graphene and CNT surfaces have been presented in Fig. 3(a) and (b) respectively.
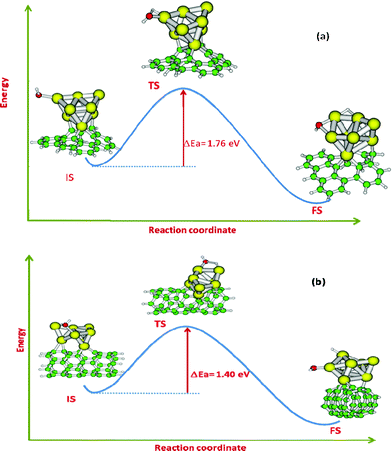 | ||
| Fig. 3 Water dissociation pathways on (a) Au8-defective graphene and (b) Au8–CNT systems. | ||
Let us first discuss the results for water dissociation on Au8 adsorbed on defective graphene. Due to the presence of a vacancy site, the adsorption energy of the cluster Au8 on the defective graphene surface is found to be −2.39 eV which is significantly higher as compared to the adsorption of the same cluster on a non-defective planar graphene (∼−0.3 eV). This signifies that the vacancy site on the surface of the graphene can, in principle, help in strong adherence of the gold nanoclusters and act as an active site for the catalysis. This strong binding also helps in significant charging of cluster Au8 (qAu8) which is found to be 0.73 in atomic units calculated from a natural population analysis. Interestingly, the binding energy of a water molecule to the cluster (IS of Fig. 3(a)) is also considerably higher (−1.42 eV) as compared to that of a gas phase Au8 cluster (−0.37 eV) discussed already. The H2O molecule prefers to bind to a low-coordinated Au-atom through an O-atom, similar to the case of the bare Au8 cluster. The optimized Au–O distance is 2.423 Å which is slightly shorter than the gas phase cluster case. The O–H bond distances (0.974 Å and 0.973 Å) and H–O–H bond angle (105.62°) for the graphene–Au8–H2O system do not vary appreciably as compared to the case of the Au8–H2O system. In the TS geometry (Fig. 3(a)), the O–H bond lengths are 0.977 Å and 1.973 Å (reaction coordinate). Also, there is significant shortening of the Au–O bond distance (2.129 Å) as compared to the corresponding IS geometry. The barrier for dissociation of water for this system is computed to be 1.76 eV. Thus, we observe that the activation barrier for water dissociation is reduced for the graphene–Au8–H2O system compared to the corresponding bare cluster. This reduction can be attributed to the significant charging of the cluster in the case of defective graphene as the substrate, and otherwise the reaction proceeds with a high activation barrier in the case of the pristine gold cluster. Let us now turn our attention to the case of water dissociation taking place on Au8 adsorbed on a defective CNT (Fig. 3(b)). The binding energy of Au8 on top of the defective CNT is −3.53 eV. This binding energy is remarkably higher than the case of defective graphene (−2.39 eV) and hence, it is expected that the CNT–gold cluster system will be highly stable under normal conditions and can act as a robust catalyst system. These two surfaces differ only by their inherent surface curvature. Our results clearly reveal that a curved surface like that of a CNT can provide better adherence for the nanoclusters and can be helpful in promoting the catalysis. The charge on the cluster Au8 is found to be 1.06 (in atomic units) which is even higher than the case of graphene–Au8. The favorable binding and charging of the cluster seem to correlate with the increase in curvature of the surface of the carbon nanostructure. A curved CNT surface can manifest stronger binding as well as charging of the clusters.
To get further insights into the favorable binding and activation of the gold cluster with the above two surfaces, we have herein presented the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of different systems, namely the pristine gold cluster, the bare surfaces and the cluster adsorbed on both the surfaces (Fig. 4) using the Chimera30 software. The HOMO and LUMO of substrates (graphene and a CNT) are mostly derived from the p-π orbitals of the carbon atoms. For the CNT–Au8 system the electronic structure of the CNT is not significantly altered which can be easily visualized from the presence of p-π orbitals in the HOMO and LUMO of the system. This infers the stability of the Au8–CNT system upon binding with the cluster. On the contrary, the presence of the p-π orbitals in the HOMO and LUMO orbitals of bare graphene is found to be significantly diminished in the frontier orbitals (HOMO and LUMO) of the graphene–Au8 system. This tends to destabilize the system. Additionally, the orbital pictures are also consistent with the charging of the gold cluster that we have mentioned previously. We can clearly notice significant charge transfer from the cluster to the substrate in the case of the Au8–CNT system as compared to the Au8–graphene system. This highly cationic gold cluster is found to favorably assist the water dissociation reaction which we have emphasized in our subsequent discussion.
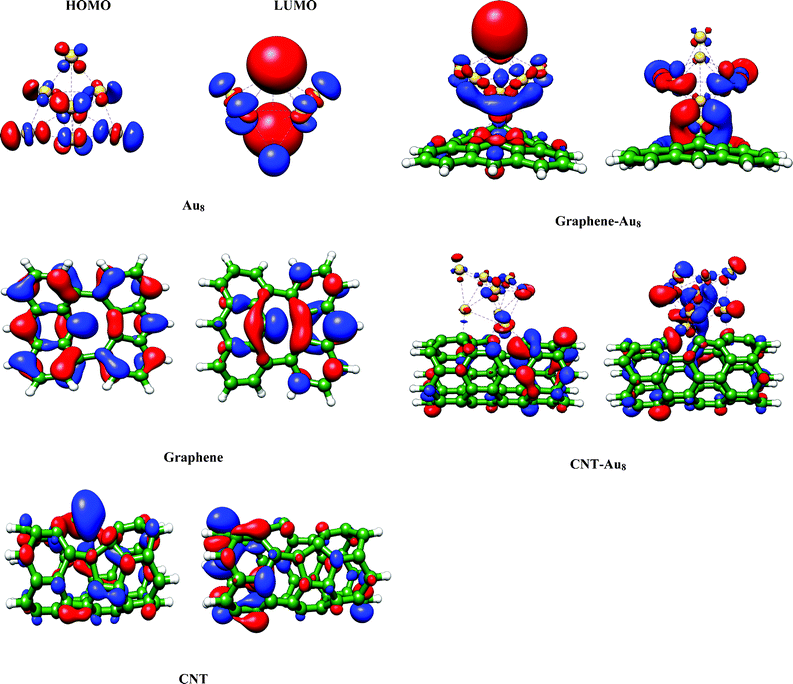 | ||
| Fig. 4 The HOMO and LUMO pictures of the bare gold cluster, bare substrates and the cluster adsorbed on the substrates (generated with an isosurface of 0.03). | ||
It is pertinent to note that some insightful molecular dynamics (MD) studies31 in recent years have unequivocally demonstrated the stability of gold clusters at finite temperatures. These results can be useful in designing stable and robust nanostructures based on gold nanoclusters for their practical applications. For instance, De et al.31 have recently studied the stability of the Au8 cluster using the Born–Oppenheimer MD simulations method and their results reveal that there is a distortion of the surface atoms of the cluster at 400 K while the central atoms are more rigid and remain very close to their equilibrium positions. They have further asserted that the cluster undergoes a solid-like to a liquid-like structure transformation above 900 K. Hence, we also expect that our complex Au8–CNT system should be stable enough for practical purposes as the melting point of a CNT is above 2000 K.32 In another recent study by Pal and co-workers,33 the reactivity properties of gold clusters using the density based reactivity descriptors have been analyzed and their results show that the size and shape of the cluster determines the reactivity of the each atomic site in the cluster which predominantly depends on the planar or non-planar conformations of the cluster structure. Such information can supplement in describing the reactivity of nanostructures of gold.
We will now resume our discussions on the dissociation of water on the Au8–CNT system. In the IS geometry, the Au–O bond distance is 2.433 Å, O–H bond distances are 0.993 Å and 0.975 Å, and the H–O–H bond angle is 104.19°. The binding energy of a water molecule to the gold cluster in this case is found to be −0.44 eV. In the optimized TS geometry, the Au–O bond length is shortened to 2.079 Å from the initial value of 2.433 Å. The O–H bond distances are 0.976 Å and 1.979 Å (reaction coordinate). The barrier for water dissociation in this case is computed to be 1.40 eV. This barrier height is less than the previous two cases, which were the pristine gold cluster and the graphene–gold cluster system.
We can attribute the facile dissociation of water on Au8 supported on a CNT on the basis of charging of the gold cluster. It is pertinent to mention that the charging effect of Au-clusters plays an extremely important role on their catalytic properties. In the context of CO oxidation promoted by nanoscale gold, it has been observed by the seminal work of Yoon et al. that the charging effects are of paramount importance to the effective bonding and oxidation of CO on Au8 supported on MgO.34 In one of our recent studies,28 we have also demonstrated the enhancement of reactivity of the gold cluster towards the CO molecule for the cases where the negative charge density is induced at the reaction center due to dopant atoms, especially hydrogen atoms. For the present water molecule dissociation process, we observe that the bare gold cluster, Au8 consists of two distinct types of atoms, similar to a core–shell cluster, having four atoms in each region. The gold atoms belonging to the inner core and outer shell regions are found to have negative and positive charges, respectively. This type of description is maintained even for the cases of graphene and CNT complexes with a marginal change in the structure. Since the water molecule is interacting with the gold cluster through the oxygen atom, the binding site with most positive charge is highly preferred to the other regions. Such active centers with a highly positive charge are observed in all cases and the observed order is CNT–Au8>graphene–Au8>Au8. Hence, it is expected that the water dissociation process should be more facile with the CNT–Au8 system than the corresponding graphene and bare gold cluster cases. This explanation corroborates with the observed trend in the reaction barriers of the three different systems towards the water dissociation. The larger perspective that we incur from the above discussions is that the cationic gold centers are more reactive towards the water dissociation process and suitably choosing substrates like the CNT, the charging effects of the cluster can be actively modulated for better catalysis.
Based on the encouraging observation of facile dissociation of a single water molecule on the Au8–CNT system, we looked ahead to consider the subsequent dissociation of a second water molecule on this system. The optimized geometry of the second water molecule adsorbed on the FS of the previous step is presented in Fig. 5 (IS). The binding energy of this water molecule is found to be −0.46 eV. This tells us that the second water molecule can also favorably bind with the product state of the first water dissociation step. The barrier for water dissociation, in this case, is calculated to be 1.12 eV. This barrier is even less than that of the first water molecule on this system. Interestingly, the product of this step (FS in Fig. 5) leads to generation of a H2 molecule. To view these results in a broader perspective, we can infer that the above system can be an ideal nanostructure where facile dissociation of two water molecules in subsequent steps can lead to the formation of molecular hydrogen.
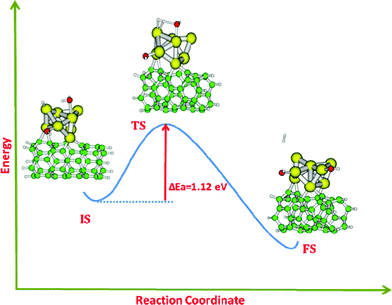 | ||
| Fig. 5 Dissociation pathway of the second water molecule and the generation of H2 on the Au8–CNT system. | ||
The important conclusion we derive from the preceding discussions is that, while water dissociation on a bare gold cluster proceeds through a higher activation barrier, the barrier can be significantly reduced by considering suitable substrates. Defective graphene and CNTs can be good choices for the substrate. The latter are a better candidate in terms of providing stronger binding of the cluster to the surface as well as significantly reducing the barrier height. In addition, we have demonstrated the effect of the substrate in tuning the efficiency of water dissociation on the gold cluster where charging of the cluster can be of great relevance. It is also worthwhile to mention that the electronic properties of small gold clusters are very much size dependent. Hence, several possibilities like proper choice of nanocluster size, choice of substrate, presence of defects on the surface are of paramount importance for the design of nanostructures for effective water dissociation.
4. Conclusions
In the present investigation, we have considered water dissociation on a bare as well as a supported gold cluster Au8. Two important carbon nanostructures namely graphene and a CNT have been chosen as the substrate surfaces. These two surfaces have defects on them in the form of a single vacancy site. It is observed that the water dissociation on the pristine gold cluster is a process with a higher barrier of 2.04 eV. This barrier height, however, is reduced to 1.76 and 1.40 eV for graphene–Au8 and CNT–Au8 systems, respectively. In addition, we have also demonstrated the subsequent dissociation of a second water molecule on the CNT–Au8 system which proceeds with a lower barrier of 1.12 eV. The generation of a H2 molecule is also worth mentioning as a product of the final step for the CNT–Au8 system. The vacancy on the surface of graphene/CNT provides stronger adherence to the cluster. The binding energy of the cluster on the curved CNT surface is more than that on the planar graphene surface, indicating the CNT to be a better choice for the surface. The charging of the cluster on these two surfaces plays a key role in deciding the catalytic efficiency. It is proposed that CNT decorated gold clusters can be a potential catalyst for water dissociation and concomitant liberation of molecular hydrogen. Factors, such as the nature of the CNT, shape and size of gold clusters can be influential in fine-tuning the efficiency of these nanostructures for water dissociation. Our study projects a step beyond the conventional realm of catalysis by nanoscale gold which has been explored in great detail for different kinds of oxidation reactions, by demonstrating its potential importance as a catalyst for water dissociation and this study is likely to stimulate further interest in this direction.Acknowledgements
N. K. J gratefully acknowledges the Homi Bhabha National Institute (HBNI), Department of Atomic Energy (DAE)-India for a senior research fellowship. The authors also acknowledge the Anupam Super computers developed by the Computer Division of BARC for providing a high performance parallel computing facility. S. K. G acknowledges DST-India for a Sir J. C Bose fellowship and also support from the INDO-EU project MONAMI.References
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1 CrossRef CAS.
- P. A. Thiel and T. E. Madey, Surf. Sci. Rep., 1987, 7, 211 CrossRef CAS.
- A. A. Phatak, W. N. Delgass, F. H. Ribeiro and W. F. Schneider, J. Phys. Chem. C, 2009, 113, 7269 CAS.
- Y. Lei, Z. X. Guo, W. Zhu, S. Meng and Z. Zhang, Appl. Phys. Lett., 2007, 91, 161906 CrossRef.
- M. Pozzo, G. Carlini, R. Rosei and D. Alfe, J. Chem. Phys., 2007, 126, 164706 CrossRef.
- Y. Liu, L. Huang, K. E. Gubbins and M. B. Nardelli, J. Chem. Phys., 2010, 133, 084510 CrossRef.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Noskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493 CrossRef CAS.
- G. C. Bond and D. T. Thompson, Catal. Rev. Sci. Eng., 1999, 41, 319 CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301 CrossRef CAS.
- A. Funakawa, I. Yamanaka, S. Takenaka and K. Otsuka, J. Am. Chem. Soc., 2004, 126, 5346 CrossRef CAS.
- W. C. Ketchie, Y. L. Fang, M. S. Wong, M. Murayama and R. J. Davis, J. Catal., 2007, 250, 94 CrossRef CAS.
- B. Xu, X. Liu, J. Haubrich and C. M. Friend, Nat. Chem., 2010, 2, 61 CrossRef CAS.
- G. E. Johnson, R. Mitric, V. Bonacic-Koutecky and A. W. Castleman Jr, Chem. Phys. Lett., 2009, 475, 1 CrossRef CAS.
- L. M. Molina, A. Lesarri and J. A. Alonso, Chem. Phys. Lett., 2009, 468, 201 CrossRef CAS.
- H. Tsunoyama, H. Sakurai and T. Tsukuda, Chem. Phys. Lett., 2006, 429, 528 CrossRef CAS.
- M. Date, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129 CrossRef CAS.
- S. D. Senanayake, D. Stacchiola, P. Liu, C. B. Mullins, J. Hrbek and J. A. Rodriguez, J. Phys. Chem. C, 2009, 113, 19536 CAS.
- R. A. Ojifinni, N. S. Froemming, J. Gong, M. Pan, T. S. Kim, J. M. White, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2008, 130, 6801 CrossRef CAS.
- X.-J. Kuang, X.-Q. Wang and G.-B. Liu, J. Mol. Model., 2011, 17, 2005 CrossRef CAS.
- K. S. Novoselov, Nat. Phys., 2009, 5, 862 CrossRef CAS.
- K. S. Novoselov, Electrochem. Soc. Int., 2011, 20, 45 CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- R. Ahlrichs, M. Bar, M. Hoser, H. Horn and C. Kolmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- R. M. Olson, S. Varganov, M. S. Gordon, H. Metiu, S. Chretien, P. Piecuch, K. Kowalski, S. A. Kucharski and M. Musial, J. Am. Chem. Soc., 2005, 127, 1049 CrossRef CAS.
- N. K. Jena, K. R. S. Chandrakumar and S. K. Ghosh, J. Phys. Chem. Lett., 2011, 2, 1476 CrossRef CAS.
- D. R. Kauffman, D. C. Sorescu, D. P. Schofield, B. L. Allen, K. D. Jordan and A. Star, Nano Lett., 2010, 10, 958 CrossRef CAS.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605 CrossRef CAS.
- H. S. De, S. Krishnamurty, D. Mishra and S. Pal, J. Phys. Chem. C, 2011, 115, 17278 CAS.
- M. S. Dresselhaus, G. Dresselhaus and P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, New York, 1996 Search PubMed.
- H. S. De, S. Krishnamurty and S. Pal, J. Phys. Chem. C, 2010, 114, 6690 Search PubMed.
- B. Yoon, H. Hakkinen, U. Landman, A. S. Worz, J. M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |