DOI:
10.1039/C2RA21039H
(Paper)
RSC Adv., 2012,
2, 9952-9957
New self-assembled supramolecular polymers formed by self-complementary sextuple hydrogen bond motifs†
Received
25th May 2012
, Accepted 19th August 2012
First published on 20th August 2012
Abstract
A new supramolecular material, N-(6-(3-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)propanamido)pyridin-2-yl)undec-10-enamide (U-DPy) possesses an extremely high association constant (Ka >107 M−1), which is able to dimerize through a self-complementary sextuple hydrogen-bonding array. As the end groups of an oligomer, U-DPy forms a self-assembling polymer system. These new mono- and difunctional telechelic supramolecular polymers have highly prominent properties, leading to a substantial increase in viscosity, thermal stability and unique morphology changes in the solid state. More surprisingly, they can self-organize into lamellar crystallization and a spherical structure. In addition, upon increasing the concentration in the solution state, the size of the resulting spherical structure is gradually increased. These spherical structures are constructed from supramolecular polymers with a high degree of polymerization.
Introduction
Supramolecular materials permit a rapid response to a change in environment, resulting in easily controllable strength and selectivity of noncovalent interactions. A typical example is the assembling and disassembling1 of structural motifs such as DNA—fundamental for translation and transcription in biology. Formation of polymers or polymer networks via noncovalent interactions is therefore an attractive approach to constructing stimuli-responsive soft materials.2,3 Recently, supramolecular chemistry has evolved to a stage where the paradigm can be explored, with building blocks now widely available that (a) are easy to make in multi-gram quantities and (b) interact in a defined manner with sufficient strength.
Since Carothers et al. established his influential theory on polymerization 81 years ago,4 there have been numerous synthetic approaches to obtain high-molecular-weight polymers through condensation polymerization.5 Condensation polymers all have two properties in common—staticity and nonreversibility, either in the polymerization process or in the cross-linking process. Indeed, sequentially reversible polymers are difficult to synthesize through covalent bonds, but similar architectures can be achieved through strong noncovalent interactions, utilizing highly complementary noncovalent bonds.6 To these polymers, the simplest structure is derived from hydrogen-bonded polymers in which the proton donor and acceptor groups are located in separate oligomer main chains or chain ends, allowing the new supramolecular polymer to be produced without covalent polymerization.7 Recently, several supramolecular structures have been incorporated into polymers to form materials with the features of conventional polymers and reversibility in the bonding between monomer units.8 By choosing the appropriate hydrogen bonding motif, strength of bonding with a binding constant larger than 106 M−1 can be obtained, which is close to the strength of conventional covalent linkages.1,7 Meijer and co-workers9,10 established a series of supramolecular polymers exhibiting mechanical properties that were thought to be reachable only by covalently linked monomers in macromolecules. In addition, Lehn11 and Binder12 used the hydrogen bonding motif for preparing chain-extended supramolecular polymers and block copolymers, which can assemble via a strong six-point hydrogen bond association. Nevertheless, it is still a challenge to control the non-covalent polymers with secondary (and higher) bonding from different hydrogen-bonding motifs.13,14
Our recent findings15 indicate that a new derivative of N-(6-(3-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)propanamido)pyridin-2-yl)undec-10-enamide (U-DPy) can dimerize easily and strongly by means of a self-complementary DADADA (donor–acceptor–donor–acceptor–donor–acceptor) array of sextuple hydrogen bonding interactions. 1H NMR titration studies in CDCl3 showed that the self-complementary complexes formed rapidly on the NMR time scale with extremely high association constants (Kdimer > 107 M−1). More surprisingly, the strength of hydrogen bonding formed through dimerization was over the fluorescence time scale, prompting us to this functionality as an end group in reversible self-assembling polymer systems. To the best of our knowledge, this is the first example of the formation of a U-DPy-based supramolecular polymer in which the new polymer possesses polymeric properties in the solution as well as in the bulk state.
Results and discussion
Synthesis and viscosity behavior of the U-DPy telechelic supramolecular polymer
We present here a general route for preparing well-defined, end-functional U-DPy polymers of controlled molecular architectures, attainable through supramolecular polymerization from strongly associating U-DPy functional groups. Polyethylene glycol (PEG) was chosen as the polymer backbone due to its wide application areas in the chemical, biomedical, and industrial fields. Two low molecular weight oligomers, mono- and dipropargyl terminated PEG 1000, were functionalized by the click reaction with azide-labeled U-DPy (N3-U-DPy) in high yields (>90%). The products are U-DPy-PEG1T and BU-DPy-PEG1T, whose structures are shown in Fig. 1a (detailed synthetic strategy is described in the ESI†). Most importantly, the variable-temperature NMR (VT-NMR) experiments of MU-DPy-PEG1T and BU-DPy-PEG1T in tetrachloroethane-d2 (shown in Fig. S5a and S5b, ESI†) obtained at 30 °C show a clear downfield peak (12.61 ppm) for the N–H proton (of the uracil units), which corresponds to the formation of the selective hydrogen bonded complex between uracil and diamidopyridine (DAP).15 When these samples were heated from 30 to 120 °C, the peak of the amide proton was shifted slightly upfield from 12.61 to 11.91 ppm. This indicates that the hydrogen bonding interaction is not appreciably destroyed during heating, which was rarely observed. In these previous studies, the multiple hydrogen bonding systems were significantly influenced by an increase in temperature, showing an obvious shift of amide proton signals.16,17 In addition, we also performed VT-NMR on azide-labeled U-DPy (a precursor for the synthesis of U-DPy-modified polymers) and a similar trend was seen for MU-DPy-PEG1T and BU-DPy-PEG1T (Fig. S6†). This observation further indicates that the U-DPy PEG system forms highly stable hydrogen-bonded complexes in tetrachloroethane. To further evaluate the effect of hydrogen-bonding associations on solution viscosity, the viscosities of MU-DPy-PEG1T and BU-DPy-PEG1T in chloroform at ambient temperature were measured using an Ubbelohde viscometer (Fig. 1b). The viscosity of the BU-DPy-PEG1T increased nonlinearly with concentration. When the concentration is below 66 mg dL−1, the slope of the line is calculated as 0.0917, while above the critical concentration the slope becomes 0.46. This result indicates that the concentration dependent association between the individual monomer units is significantly greater than both PEG 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and MU-DPy-PEG1T, even though the PEG 20000 has a higher molecular weight with more entanglements. These results from BU-DPy-PEG1T may come from the equilibrium between the linear polymeric and oligomeric cyclic structures (Scheme 1). In other words, raising the BU-DPy-PEG1T concentration induces the transition from the kinetically favorable intra-molecular interaction (at low concentration) to the dynamically favorable intermolecular hydrogen bonding interaction (at high concentration) and results in an increase in the degree of polymerization.10
000 and MU-DPy-PEG1T, even though the PEG 20000 has a higher molecular weight with more entanglements. These results from BU-DPy-PEG1T may come from the equilibrium between the linear polymeric and oligomeric cyclic structures (Scheme 1). In other words, raising the BU-DPy-PEG1T concentration induces the transition from the kinetically favorable intra-molecular interaction (at low concentration) to the dynamically favorable intermolecular hydrogen bonding interaction (at high concentration) and results in an increase in the degree of polymerization.10
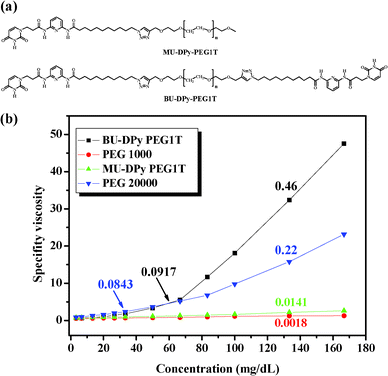 |
| | Fig. 1 (a) Chemical structures of the U-DPy telechelic supramolecular polymer. (b) Specific viscosity of MU-DPy-PEG1T, BU-DPy-PEG1T and the control PEG versus concentration in CHCl3 at 25 °C. | |
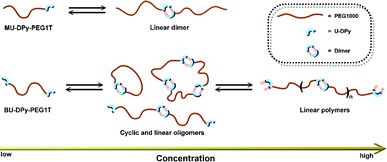 |
| | Scheme 1 Schematic representation of the supramolecular polymerization obtained from mono- and difunctional U-DPy derivatives, respectively, as a function of concentration. | |
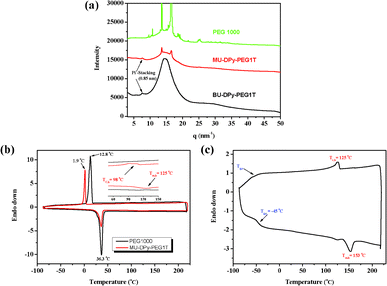 |
| | Fig. 2 (a) WAXS data for annealing MU-DPy-PEG1T, BU-DPy-PEG1T and the control PEG 1000. DSC curves of (b) MU-DPy-PEG1T and (c) BU-DPy-PEG1T. | |
Interestingly, the MU-DPy-PEG1T shows a lower crystallization peak (Tc = 1.9 °C) than the control PEG 1000 (Tc = 12.8 °C), probably due to the association of certain units affecting the crystallization behavior.19 This intriguing behavior leads us to investigate microstructures of this phenomenon in more detail through isothermal crystallization kinetics and atomic force microscopy (AFM) measurements. The typical isotherm data (Fig. 3a) of MU-DPy-PEG1T and control PEG 1000 indicate that introducing a U-DPy end group results in a significant reduction of the overall kinetic rate constant (K), and the crystallization rate is progressively decreased. In addition, the Avrami exponents (n) of MU-DPy-PEG1T are around 2.1–2.5, which is higher than PEG 1000. This result reveals the presence of two-dimensional crystallization20 and stacks of dimerized U-DPy moieties affecting the crystallization morphology. To further confirm the isotherm results, we performed an AFM experiment to visually observe the crystal structure of MU-DPy-PEG1T. As shown in Fig. 3b, the PEG 1000 exhibits a typical spherulite morphology. In contrast, MU-DPy-PEG1T forms lamellar objects instead of spherical objects (Fig. 3c), suggesting that the PEG crystals grow preferentially in some directions due to the presence of the U-DPy end group. As well as the isotherm data, the AFM images also revealed that this MU-DPy-PEG1T system exhibits the strongest segregation strength and results in a stable and confined crystallization in the lamellar arrangement.
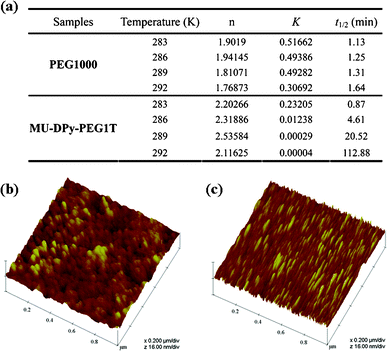 |
| | Fig. 3 (a) The table presents the kinetic data and (b, c) AFM images showing the spherical and lamellar crystal morphology of annealed (b) PEG 1000 and (c) MU-DPy-PEG1T, respectively. | |
Self-assembly of BU-DPy-PEG1T in the bulk state
Supramolecular self-assembly can be used as a bottom-up approach for the development of advanced structures.21 If these hydrogen bonds are strong enough, supramolecular polymerization can refer to the objects directly.22 We performed variable-temperature small-angle X-ray scattering (SAXS) and atomic force microscopy (AFM) to understand the self-assembly behavior of BU-DPy-PEG1T. Fig. 4a displays the SAXS profiles of BU-DPy-PEG1T at 25 °C. Surprisingly, annealing BU-DPy-PEG1T resulted in the appearance of an ordered regularization. Highly ordered lamellar microstructures are observed (n = 1–3) with the first intense reflection at q = 1.10 nm−1 corresponding to a spacing of d = 5.71 nm. Upon heating from 25 to 150 °C, the SAXS profile remains nearly unchanged, as shown in Fig. 4a, implying that BU-DPy-PEG1T forms highly stable lamellar structures in the bulk state.23,24 In addition, the one-dimensional correlation function is employed to analyze the detailed structural information (Fig. 4b) and determine the most probable average length period (L), the average crystalline lamellar thickness (L1) and the average interlamellar amorphous layer thickness (L2). Based on the self-correlation triangle method, the mean thickness of L1 is 1.51 nm and the mean thickness of L2 from L2 = L − L1 (5.71 − 1.51 = 4.20 nm) is 4.20 nm, which is constructed on lamellae microstructures as depicted in Scheme 2. Accordingly, the lamellar structures are formed by the PEG region of 4.2 nm in length, connected side by side with the dimerized U-DPy moieties (1.51 nm). In addition, the close π-packing of these lamellae with a d spacing of 0.85 nm (Fig. 2a) also facilitates the system to form stable structures. On other hand, spherical particles were observed from AFM with samples prepared by dropping chloroform solutions of BU-DPy-PEG1T in different concentrations onto wafer surfaces, followed by drying and annealing (Fig. 5a–d). More surprisingly, the gradual increase in particle size with increasing concentration can be stabilized to maintain spherical structures throughout its growth (Fig. 5e). Remarkably, it can exist as a stable ‘zero-dimensional’ nanostructure based on these observations, rather like the previous reports found in fibrous or higher structures.18,25,26 These results also confirmed that BU-DPy-PEG1T was indeed present in unimolecular micelles. In addition, we found that sextuple hydrogen bonding alone is enough to permit assembly of the chain into a well-defined particle.
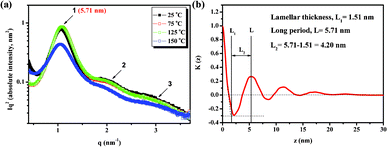 |
| | Fig. 4 (a) One-dimensional SAXS data and (b) one-dimensional correlation function calculated from the SAXS data for BU-DPy-PEG1T recorded at room temperature. | |
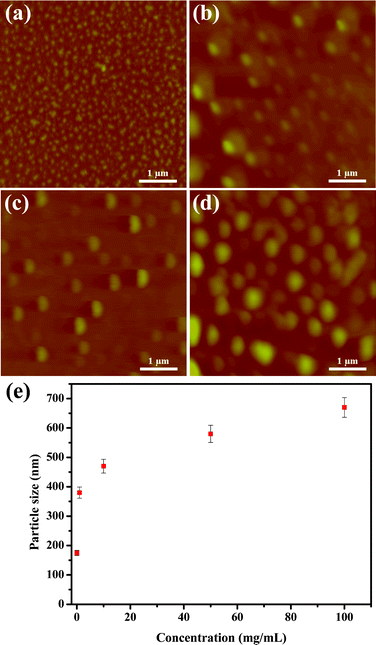 |
| | Fig. 5 AFM images of annealed BU-DPy-PEG1T on the wafer surface with different concentrations: (a) 0.0067, (b) 1, (c) 10 and (d) 100 mg mL−1. (e) Particle size of annealed BU-DPy-PEG1T versus concentration as measured by AFM. | |
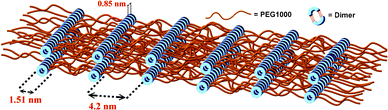 |
| | Scheme 2 Graphical representations of the lamellar structures of the BU-DPy-PEG1T self-complexes in the bulk state. | |
To further evaluate the propensity of the new module for spherical growth, diffusion coefficient (D) measurements in 16.7 and 167 mg dL−1 solutions of BU-DPy-PEG1T in CDCl3 were undertaken in order to analyze the degree of self-association in different concentrations. For the solution with a concentration of 16.7 mg dL−1, the diffusion coefficient is calculated to be 2.35 × 10−10 m2 s−1, indicating the presence of cyclic oligomers in dilute chloroform solutions.10 Further polymerization occurs upon increasing the concentration, as confirmed by an extremely slow diffusion, with a D value of 9.81 × 10−12 m2 s−1 for 167 mg dL−1, implying a high degree of polymerization for BU-DPy-PEG1T. In addition, the DP of the supramolecular polymer was calculated to be approximately 146 at a concentration of 167 mg dL−1.27 Diffusion coefficient measurements also agree well with the viscosity results (Fig. 1b).
With these data in hand, the growth mechanism of the spherical structure can be easily explained via the Flory–Huggins theory.28 This theory predicts the transition of cylindrical aggregation to micellar aggregation with an increase in the volume fraction of spherical aggregation.29 In other words, the morphology from zero-dimensional growth at very dilute concentrations can be attributed to the clustering of cyclic oligomers30 and the formation of small particles (Fig. 5a and Scheme 3). In contrast, large particles formed at high concentration—presumably by clustering of linear polymers31 (Fig. 5d and Scheme 3), which constructed supramolecular polymers with a high degree of polymerization. These results also demonstrate the morphological nature of the collapsed particle state. As observed by Brus and coworkers, the self-assembled morphologies can be modeled with increasing domain size on the surface, and are in good agreement with our results.32 Ultimately, this work presents a new phenomenon to an elusive target: incorporation of D-UPy units into polymers can be turned into a high-performance supramolecular material.
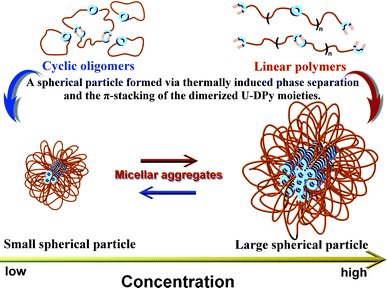 |
| | Scheme 3 Suggested processes of the formation of the micellar aggregations in the bulk state. | |
Conclusions
In summary, we have demonstrated that the recognition units possessing extremely high Ka lead to new supramolecular polymers in the bulk as well as in solution. Incorporation of U-DPy units into PEG chain ends results in phase separation, dramatically changed morphology and formation of a supramolecular complex chain with an extremely high degree of polymerization. In addition, the U-DPy telechelic derivative possesses polymer-like properties as a result of self-complementary interactions, providing a potential route toward the design and fabrication of polymer-like supramolecular materials.
Experimental section
Materials
All other chemicals were purchased from Aldrich (USA) or Acros Organics (Germany) and were used as received. All solvents were purchased from TEDIA (USA) and distilled over CaH2 prior to use.
Syntheses of supramolecular polymers
MU-DPy-PEG1T and BU-DPy-PEG1T were prepared by “click chemistry” of U-DPy azide in the presence of propargyl PEG 1000 using CuBr/PMDETA as the catalyst system. The general materials in this work are described in more detail in the ESI.†
Characterizations
Viscosity experiment.
The effects of hydrogen-bonding associations on solution viscosities of MU-DPy-PEG1T and BU-DPy-PEG1T in CHCl3 solution at ambient temperature were measured using an Ubbelohde viscometer.
Nuclear magnetic resonance (NMR) measurement.
1H-NMR spectra were recorded using a Varian Inova-500 MHz spectrometer equipped with a 9.395 T Bruker magnet. Samples of ca. 5 mg were analyzed at 25 °C in deuterated solvent. 13C-NMR spectra were obtained on a Varian Inova- 500 MHz spectrometer operated at 125 MHz. All samples of ca. 20 mg were dissolved in deuterated solvent and analyzed at 25 °C.
Elemental analysis (EA).
The carbon, hydrogen, and nitrogen contents of the samples were obtained using a CHN-O-Rapid elemental analyzer (Foss. Heraeus, Germany).
Dynamic light scattering (DLS).
DLS measurements were conducted using a 90Plus laser particle size analyzer (Brookhaven Instruments Corp., USA), which was calibrated using a 60 nm latex standard. The scattering from a small amount of the sample dissolved in chloroform was measured at 90°. The hydrodynamic radius (r) of the aggregates was calculated using the Stokes–Einstein equation: r = kBT/6πηD, where kB is the Boltzmann constant, T is the absolute temperature, η is the solvent viscosity, and D is the diffusion constant.
FT-IR spectroscopy.
Recorded using a Nicolet Avatar 320 FT-IR spectrometer; 32 scans were collected at a spectral resolution of 1 cm−1. The conventional KBr disk method was employed: the sample was dissolved in chloroform and then cast onto a KBr disk and dried under vacuum at 70 °C.
Polarized optical microscopy.
The cover glass containing the sample film was placed under a polarized optical microscope (Olympus BX51) equipped with a camera system.
Differential scanning calorimetry (DSC).
DSC was performed using a TA DSC-Q20 controller operated under a dry nitrogen atmosphere. The samples were weighed (ca. 5–10 mg), sealed in an aluminum pan, and then heated and cooled from −90 to +240 °C at a rate of 10 °C min−1.
Wide angle X-ray scattering (WAXS).
WAXS was collected by the 01C2 SWLS beamline of the National Synchrotron Radiation Research Center (NSRRC), Taiwan. For the experiment, radiation with a wavelength of 0.103316 nm was used. The beam possessed a diameter of 100 mm at the sample position, which was defined by a pinhole collimator. All tests were performed at ambient conditions (23 °C, 65% relative humidity).
Small-angle X-ray scattering (SAXS).
SAXS data were collected using the BL17A1 wiggler beamline of the National Synchrotron Radiation Research Center (NSRRC), Taiwan. The samples were sealed between two Kapton windows (thickness: 12 μm) and measured at room temperature. An X-ray beam with a diameter of 0.5 mm and a wavelength (λ) of 1.1273 Å was used for the SAXS measurement (Q range: 0.015–0.3 Å−1). The Q values of the SAXS profiles were calibrated using a polyethylene standard, Ag behenate, and tripalmitat.
Atomic force microscopy (AFM).
Height measurements of the microcapsules were determined using tapping-mode AFM (Digital Instrument NS4/D3100CL/Multi- Mode AFM; Veeco-Digital Instruments, Santa Barbara, CA) with silicon cantilevers (Pointprobe Silicon AFM Probe) at 25 °C in air. The samples for AFM investigation were prepared by placing a drop of the sample solution onto a wafer substrate and then evaporating the solvent (chloroform).
Diffusion NMR experiments.
Diffusion coefficients were recorded on a Bruker NMR spectrometer, AMX400. Full details of diffusion NMR experiments (all at 298 K) were performed as in the procedures described previously.33 A convection compensated pulse sequence was used in this work.34
Acknowledgements
We thank National Synchrotron Radiation Research Center (NSRRC, Taiwan) for the support in the SAXS measurement. This study was supported financially by the National Science Council, Taiwan (contract no. NSC 99-2120-M-009-008).
References
- D. Philp and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1996, 35, 1155 CrossRef CAS
 .
.
-
A. Ciferri, Supramolecular Polymers, 2nd edn, Taylor and Francis, Boca Raton, Florida, 2005 Search PubMed .
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS .
- W. H. Carothers, Chem. Rev., 1931, 8, 353 CrossRef CAS .
-
G. Odian, Principles of Polymerization, Wiley, New York, 1991 Search PubMed .
- J.-M. Lehn, Aust. J. Chem., 2010, 63, 611 CrossRef CAS .
- S. Sivakova and J. Rowan, Chem. Soc. Rev., 2005, 34, 9 RSC .
- J. D. Fox and S. J. Rowan, Macromolecules, 2009, 42, 6823 CrossRef CAS .
- O. A. Scherman, G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11850 CrossRef CAS .
- O. A. Scherman, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 2072 CrossRef CAS .
- E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519 RSC .
- W. H. Binder, S. Bernstorff, C. Kluger, L. Petraru and M. J. Kunz, Adv. Mater., 2005, 17, 2824 CrossRef CAS .
- M. Muthukumar, C. K. Ober and E. L. Thomas, Science, 1997, 277, 1225 CrossRef CAS .
- B. Gole, S. Shanmugaraju, A. K. Bar and P. S. Mukherjee, Chem. Commun., 2011, 10046 RSC .
- C. C. Cheng, Y. C. Yen and F. C. Chang, RSC Adv., 2011, 1, 1190 RSC .
- E. Kolomiets, E. Buhler, S. J. Candau and J.-M. Lehn, Macromolecules, 2006, 39, 1173 CrossRef CAS .
- I. H. Lin, C. C. Cheng, Y. C. Yen and F. C. Chang, Macromolecules, 2010, 43, 1245 CrossRef CAS .
- N. E. Botterhuis, D. J. M. van Beek, G. M. L. van Gemert, A. W. Bosman and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3877 CrossRef CAS .
- C. C. Cheng, C. F. Huang, Y. C. Yen and F. C. Chang, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6416 CrossRef CAS .
- S. H. M. Söntjens, R. A. E. Renken, G. M. L. van Gemert, T. A. P. Engels, A. W. Bosman, H. M. Janssen, L. E. Govaert and F. P. T. Baaijens, Macromolecules, 2008, 41, 5703 CrossRef .
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS .
- G. ten Brinke, J. Ruokolainen and O. Ikkala, Adv. Polym. Sci., 2007, 207, 113 CrossRef CAS .
- B. Gong, W. Kim and C. Y. Ryu, Angew. Chem., Int. Ed., 2004, 43, 6471 CrossRef .
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13986 CrossRef CAS .
- H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265 CrossRef CAS .
- P. Y. W. Dankers, Z. Zhang, E. Wisse, D. W. Grijpma, R. P. Sijbesma, J. Feijen and E. W. Meijer, Macromolecules, 2006, 39, 8763 CrossRef CAS .
- A. Chen, D. Wu and C. S. Johnson Jr., J. Am. Chem. Soc., 1995, 117, 7965 CrossRef CAS .
- P. J. Flory, J. Chem. Phys., 1949, 17, 223 CrossRef CAS .
- F. S. Bates and G. H. Fredrickson, Annu. Rev. Phys. Chem., 1990, 41, 525 CrossRef CAS .
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS .
- V. G. H. Lafitte, A. E. Aliev, P. N. Horton, M. B. Hursthouse, K. Bala, P. Golding and H. C. Hailes, J. Am. Chem. Soc., 2006, 128, 6544 CrossRef CAS .
- E. Rabani, D. R. Reichman, P. L. Geissler and L. E. Brus, Nature, 2003, 426, 271 CrossRef CAS .
- C. E. Atkinson, A. E. Aliev and W. B. Motherwell, Chem.–Eur. J., 2003, 9, 1714 CrossRef CAS .
- A. Jerschow and N. Müller, J. Magn. Reson., 1997, 125, 372 CrossRef CAS .
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21039h |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.