An efficient and regioselective synthetic approach towards fluorinated quinolinylphosphonates†
Blazej
Duda
a,
Sergey N.
Tverdomed
*ab and
Gerd-Volker
Röschenthaler
b
aSchool of Engineering and Science, Jacobs University Bremen, Campus Ring 1, 28759, Bremen, Germany. E-mail: s.tverdomed@jacosb-university.de; g.roeschenthaler@jacobs-university.de; Fax: +49 421 200 3229; Tel: +49 421 200 3138
bDepartment of Organic Chemistry, St. Petersburg Institute of Technology (Technical University), Moskovskii Pr. 26, 190013 St., Petersburg, Russia
First published on 28th August 2012
Abstract
A synthesis of quinolines substituted by both CF2 and phosphonate groups at the 2- and 3-positions respectively, has been successfully described. In particular, the regioselective heterocyclisation of appropriate (2-cyclohexyliminomethyl)anilines with CF2X-containing alkynylphosphonates promoted by K2CO3 was carried out in dry toluene under reflux. Substituents influenced this reaction either electronically or sterically.
Introduction
The quinoline core has been an important target for organic synthesis since it appears in various molecules with enhanced biological activity.1 Such compounds frequently show anti-malaria, anti-cancer, anti-inflammatory, anti-fungal and also anti-HIV properties.2 Thus, quinine A is a common anti-malarial agent isolated from cinchona tree bark in 1820 by Pelletier.3 Camptothecin B being often used as an anti-cancer agent has also exhibited a significant anti-retroviral activity at dose levels that are well tolerated by cells.4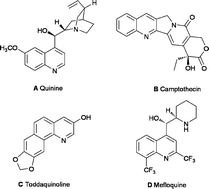
In 1993 Chen et al. isolated benzo[h]quinoline (Toddaquinoline) C which is a constituent of many Asian folk medicines.5 Mefloquine D substituted by fluorinated groups, on the other hand, developed in the 1970s, is an orally administered medication for the treatment and prevention of malaria.6
The role of incorporating phosphonate and fluorinated groups into organic compounds (aromatic and/or heteroaromatic) to alter their pharmacological properties has been well documented in the literature.7 Particularly, the phosphonate group as phosphate mimics has proved to be valuable in numerous applications.7c,8 On the other side, fluorine uniquely affects the properties of organic compounds, for instance an introduction of this atom/group into pharmaceuticals can provide better bioavailability, lipophilicity and metabolic stability, thus increasing the strength of a compound's interaction with protein.7d Therefore, incorporating fluorine-containing groups into organophosphonates is indeed very desirable.
The syntheses of the quinoline ring have been usually performed via Friedlander,9 Combes,10 Skraup,11 Doebner-von Miller,12 Conrad-Limpach,13 and Pfitzinger14 methods. However, due to the fact that β-dicarbonyl substrates are applied, the possibility of synthesizing polyfunctionalised quinolines is, indeed, limited. Among them, ynones and other related systems were also used as starting materials, even though transition metal-promoted catalysis or the additional presence of an electrophile is required.15 It must be mentioned here that only a few examples regarding the heterocyclisation reaction of either symmetrical or terminal alkynes to achieve quinolines have been reported, although with poor regioselectivity.16 Nevertheless, to the best of our knowledge, there have been no reports hitherto published concerning unsymmetrical acetylenes towards the regioselective synthesis of 2,3-disubstituted quinolines, bearing –CF2X and –P(O)(OEt)2 groups, respectively. Hence, we wish to report for the first time a one-pot, efficient, straightforward, and regioselective methodology to produce fluorinated quinolinylphosphonates.
Results and discussion
Recently, we have reported the synthesis of fluorine- and phosphorus-containing azaxanthones via heterocyclisation of non-aromatic 1,2-aminoaldehydes with suitable CF2X-containing acetylenephosphonates.17a Therefore, we investigated the preparation of the desired quinolinylphosphonates by employing an ortho-aminobenzaldehyde to react with the respective alkyne 3a–e bearing –CF2X and the phosphonate groups. Due to the fact that 2-aminobenzaldehyde 1 is susceptible to self-condensation,18 we then performed a Borsche's modification to utilize more stable o-aminobenzaldehyde imines 2 (Scheme 1).19 The desired imines were successfully synthesized by the condensation of an amine with an o-nitrobenzaldehyde followed by sodium sulfide reduction.20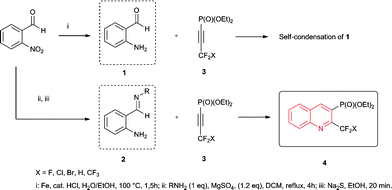 | ||
| Scheme 1 Synthetic approach towards 2,3-difuntionalized quinolines 3a–t. | ||
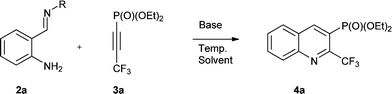
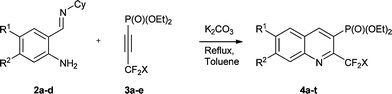
The heterocyclisation reaction of substrates 2 and 3 proceeded in the presence of a base in dry toluene under reflux to afford CF2X-containing quinolinylphosphonates 4a–t (where X = F, Cl, Br, H and CF3) in good to excellent yields (Table 2). The progress of the reaction was monitored by 19F and 31P NMR spectroscopy of the reaction mixture.
We observed that reacting 2a–d with 3a–e in the absence of a basic mediator did not occur, even at extended reaction times (entry 1, Table 1). To find optimum reaction conditions, a various mediators, such as K2CO3, NEt3, i-Pr2NEt and DABCO were employed (Table 1). These experiments were conducted by the action of a stoichiometric ratio of 2a and 3a in toluene under reflux. As a result, K2CO3 showed the best mediating ability furnishing product 4a in a 97% isolated yield (entry 2, Table 1). It should be pointed out that weaker bases, such as NEt3 and i-Pr2NEt provided 4a only in the range of 5–18% of isolated yields (entries 3–4, Table 1). Notwithstanding the very good conversion of 4 detected in the case of DABCO, the desired compound 4a was isolated in rather low yield (entry 6, Table 1), probably due to the decomposition of intermediates to an intractable material.
| Entry | Base | Solvent | R | Temp. [°C] | Convn.a [%] | Yieldb [%] |
|---|---|---|---|---|---|---|
| a Conversion was established by 19F and 31P NMR after 12h. b Isolated yields. | ||||||
| 1 | None | Toluene | Cy | 112 | — | — |
| 2 | K2CO3 | Toluene | Cy | 112 | 100 | 97 |
| 3 | K2CO3 | Toluene | Cy | 50 | 60 | 39 |
| 4 | NEt3 | Toluene | Cy | 112 | 15 | 5 |
| 5 | iPr2NEt | Toluene | Cy | 112 | 50 | 18 |
| 6 | DABCO | Toluene | Cy | 112 | 100 | 31 |
| 7 | K2CO3 | Benzene | Cy | 80 | 80 | 64 |
| 8 | K2CO3 | Acetone | Cy | 56 | 50 | 30 |
| 9 | K2CO3 | DMSO | Cy | 25 | 69 | 42 |
| 10 | K2CO3 | Toluene | Tol | 112 | 100 | 96 |
| 11 | K2CO3 | Toluene | i-Bu | 112 | 40 | 24 |
Furthermore, the role of the reaction medium, for instance toluene, benzene, acetone and DMSO upon the formation of the target products, was studied (Table 1). Regarding the conditions used, the reaction in toluene proceeded almost quantitatively (97%). Benzene, on the other hand, provided the target product 4a in only 64% isolated yield (entry 7, Table 1), whereas more polar solvents (acetone and DMSO) gave 4a in even lower yields (entries 8–9, Table 1).
In addition, we also screened a steric effect of alkyl/aryl groups adjacent to an azomethine moiety in 2 onto the synthetic efficiency, namely iso-butyl, cyclohexyl and p-methylphenyl. Interestingly, the cyclohexyl and p-toluidine differed only slightly in such heterocyclisations giving 4a in comparable reaction yields (entries 2 and 10, Table 1). However, iso-butyl as being a less bulky group provided 4a in a remarkably lower yield (entry 11, Table 1).
We then extended our study to investigate the scope of the process by taking differently substituted substrates 2a–d. Table 2 illustrates that the best results were observed when an electron-accepting group is connected to the aromatic ring, namely –CF32d (entries 4, 8, 12, 16 and 20, Table 2). As assumed, π-electron-donating groups, such as –OMe 2c significantly reduced the reaction yields; whereas –Cl 2b provided the target products 4 only in slightly lesser yields when compared to non-substituted 2a. Such findings can be easily explained by the increased nucleophilicity of the –NH2 group due to the presence of π-EDG (–OMe, –Cl), thus affecting the efficiency of this heterocyclisation by lowering the reaction yields.
| Entry | R1 | R2 | X | Product | Time [h] | Yielda [%] |
|---|---|---|---|---|---|---|
| a Isolated yields | ||||||
| 1 | H | H | F | 4a | 10 | 97 |
| 2 | Cl | H | F | 4b | 11 | 91 |
| 3 | OMe | OMe | F | 4c | 13 | 80 |
| 4 | H | CF3 | F | 4d | 10 | 99 |
| 5 | H | H | Cl | 4e | 11 | 90 |
| 6 | Cl | H | Cl | 4f | 11 | 81 |
| 7 | OMe | OMe | Cl | 4g | 13 | 71 |
| 8 | H | CF3 | Cl | 4h | 12 | 94 |
| 9 | H | H | Br | 4i | 12 | 88 |
| 10 | Cl | H | Br | 4j | 12 | 80 |
| 11 | OMe | OMe | Br | 4k | 14 | 71 |
| 12 | H | CF3 | Br | 4l | 11 | 93 |
| 13 | H | H | H | 4m | 13 | 60 |
| 14 | Cl | H | H | 4n | 14 | 54 |
| 15 | OMe | OMe | H | 4o | 16 | 42 |
| 16 | H | CF3 | H | 4p | 13 | 69 |
| 17 | H | H | CF3 | 4q | 14 | 65 |
| 18 | Cl | H | CF3 | 4r | 14 | 53 |
| 19 | OMe | OMe | CF3 | 4s | 15 | 40 |
| 20 | H | CF3 | CF3 | 4t | 12 | 67 |
Finally, we found out that fluorine-containing groups attached to alkyne 3 influenced this cyclisation reaction either electronically or sterically. In general, the more electrophilic the carbon atom adjacent to the –CF2X groups is, the better yields are observed. The –CF3 group of 3a with the greatest electron-withdrawing property in this series, indeed, furnished the corresponding 4a–d in very good to excellent yields (entries 1–4, Table 2). Whereas 3b (X = Cl) and 3c (X = Br) provided products 4e–h and 4i–l in lower reaction yields, when compared to 4a, respectively (entries 5–12, Table 2). The significant lowering of the reaction yields of CF2H–substituted quinolinylphosphonates 4m–p can be explained by the smaller EWG-effect (entries 13–16, Table 2). It must be pointed out that the –C2F5 group 3e due to the steric requirements resulted in compounds 4q–t in moderate yields, when compared to the –CF3 group (entries 17–20, Table 2).17
Monitoring the reaction between 2a and 3a by 19F NMR under optimized conditions (entry 2, Table 1) after 12 h showed the presence of substrate 3a (δF −51.4), the desired product 4a (δF −63.8) along with a signal around δF ≈ −66 which could belong to the non-aromatic 1,4-dihydroquinoline derivative 6. Moreover, the presence of the Michael adduct between 2a and 3a was not detected during the experiments. Encouraged by the experimental data given, we wish to propose a plausible synthetic pathway, where the base-activated –NH2 group (hydrogen-bonded complex) of an imine 5 regioselectively attacks the more electrophilic carbon of the acetylenephosphonate's triple bond via a Michael-type addition followed by a concerted cyclisation to form the thermodynamically unstable 1,4-dihydroquinoline derivative 6 which after aromatisation and elimination of the amine gives CF2X-containing quinolinylphosphonates 4a–t (Scheme 2).
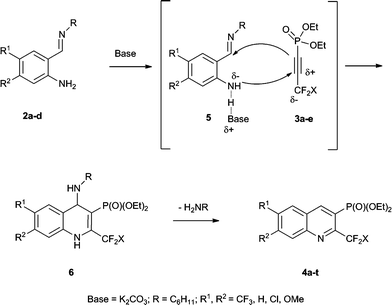 | ||
| Scheme 2 Possible synthetic pathway to CF2X-substituted quinolinylphosphonates 4a–t. | ||
The structures of the new compounds were successfully established by 1H, 13C, 19F and 31P NMR spectroscopy analysis along with high-resolution mass spectrometry. Additionally, the structure of diethyl (6,7-dimethoxy-2-(trifluoromethyl)quinolin-3-yl)phosphonate 4c21 was confirmed by single-crystal X-ray diffraction analysis (Fig. 1) and compound 4g by 2D NMR spectroscopy (13C–1H HETCOR).
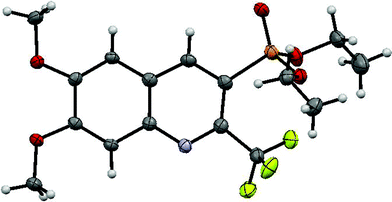 | ||
| Fig. 1 An ORTEP image of compound 4c. (Light Grey = H; Grey = C; Blue = N; Orange = P; Green = F; Red = O). | ||
In the 1H NMR, a characteristic doublet with the coupling constant 3JH–P = 16 Hz at δH ≈ 9.1 was assigned to a proton at the 4-position. The C(4) carbon atom, in the 13C NMR, emerged as a doublet at δC ≈ 147 (2JC–P = 7 Hz). The C(2) carbon atom exhibits an appropriate multiplicity, such as quartet of doublets (2JC–F = 35 Hz, 2JC–P = 8 Hz) for 4a–d and triplet of doublets (2JC–F = 29 Hz, 2JC–P = 8 Hz) for 4e–t at δC ≈ 148. The phosphorus nucleus of the phosphonate group split the carbon atom to whom is this particular group linked, to a doublet at δC ≈ 120 with 1JC–P = 186 Hz.
Conclusions
In summary, we have demonstrated a novel, straightforward, efficient and regioselective synthetic methodology involving a base-promoted heterocyclisation reaction of unsymmetrical electron-deficient fluorinated acetylenephosphonates with o-aminobenzaldehyde imines to give CF2X-containing quinolinylphosphonates in good to excellent yields. The role of reaction media and mediators as well as the effect of substituents on the aryl ring and –CF2X groups, were additionally discussed.Experimental details
General Remarks
All reagents from commercial suppliers were used without further purification. All solvents were freshly distilled before use from appropriate drying agents. All other reagents were recrystallized. Reactions were performed under an atmosphere of dry argon. Analytical TLCs were performed with silica gel 60 F254 plates. Column chromatography was carried out using silica gel 60 (230–400 mesh ASTM). Melting points were determined without correction. NMR spectra were obtained on a spectrometer operating at 400 MHz for 1H (TMS), 376 MHz for 19F (CFCl3), 161 MHz for 31P (H3PO4) and 100 MHz for 13C (TMS). All measurements were accomplished in solution in CDCl3. Mass spectrometry was established on a MicroTOF-Q fitted with an ESI source.Procedure for the Preparation of Imine Derivatives 2a–d
To the mixture of a nitrobenzaldehyde and MgSO4 in dry DCM a cyclohexylamine was added slowly. Then the suspension was refluxed for 4 h, then cooled down to ambient temperature and filtered off the remained MgSO4. The filtrate was next concentrated under reduced pressure to give pure imine derivative. Subsequently an imine was dissolved in ethanol and warmed up to 80 °C. To the hot solution solid Na2S (hydrate) was slowly added. The solution was maintained for an additional 20 min, cooled down to 0 °C and kept for 4 h. Thus formed precipitate was filtered off and the solution concentrated under reduced pressure. To the crude product, H2O was added and a new drop of precipitate was formed, filtered off, washed with H2O (3 × 100 mL) and dried to produce pure 2-(cyclohexylimino)-methyl)aniline derivatives.General Procedure for the Preparation of CF2-Containing Quinolinylphosphonates 4a–t
The mixture of 2-((cyclohexylimino)methyl)aniline derivative 2 (5 mmol) and K2CO3 (5 mmol) was dissolved in dry toluene (25 mL) at ambient temperature. To the reaction mixture an alkyne 3 (5 mmol) was charged slowly. The solution was warmed up to reflux and stirred for 10–12 h. The K2CO3 was filtered off and the remained solution concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel using DCM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc (5:1 ratio) as eluent.
:EtOAc (5:1 ratio) as eluent.
Diethyl (2-(trifluoromethyl)quinolin-3-yl)phosphonate 4a
Yellowish oil (97%); 1H NMR (400 MHz, CDCl3) δ 1.30 (t, J = 7.1 Hz, 6H), 4.15 (m, 4H), 7.67 (td, J = 8.2 Hz, J = 1.1 Hz, 1H), 7.85 (td, J = 8.2 Hz, J = 1.3 Hz, 1H), 7.92 (d, J = 8.2 Hz, 1H), 8.15 (dd, J = 8.7 Hz, J = 0.7 Hz, 1H), 9.09 (d, 3JH–P = 16.5 Hz, 1H); 13C NMR (100 MHz) δ 16.1 (d, J = 6.5 Hz), 63.1 (d, J = 5.9 Hz), 119.8 (d, 1JC–P = 186.5 Hz), 121.4 (q, 1JC–F = 275.7 Hz), 127.1 (d, 3JC–P = 12.1 Hz), 128.5, 129.6, 129.8, 132.9, 147.5 (qd, 2JC–F = 35.9 Hz, 2JC–P = 8.4 Hz), 147.6 (d, 2JC–P = 6.8 Hz); 19F NMR (376 MHz) δ −63.8; 31P NMR (161 MHz) δ 13.6; HRMS (ESI): calcd for C14H16F3NO3P [M+H]+ 334.0814, found 334.0812.Acknowledgements
B. D. acknowledges Jacobs University Bremen for a doctoral scholarship. S. N. T and G.-V. R. are thankful to the Deutsche Forschungsgemeinschaft (436 RUS 113/961/0-1) for financial support. Dr Bassem S. Bassil is thanked for the XRD data analysis.References
- (a) J. P. Michael, Nat. Prod. Rep., 2007, 24, 223 RSC; (b) J. P. Michael, Nat. Prod. Rep., 2005, 22, 627 RSC; (c) G. Roma, M. D. Braccio, G. Grossi, F. Mattioli and M. Ghia, Eur. J. Med. Chem., 2000, 35, 1021 CrossRef CAS; (d) Y.-L. Chen, K.-C. Fang, J.-Y. Sheu, S.-L. Hsu and C.-C. Tzeng, J. Med. Chem., 2001, 44, 2374 CrossRef CAS.
- (a) S. Kumar, S. Bawa and H. Gupta, Mini-Rev. Med. Chem., 2009, 14, 1648 CrossRef; (b) R. Musiol, M. Serda, S. Hensel-Bielowska and J. Polanski, Curr. Med. Chem., 2010, 17, 1960 CrossRef CAS; (c) K. Kaur, M. Jain, P. R. Reddy and R. Jain, Eur. J. Med. Chem., 2010, 45, 3245 CrossRef CAS; (d) R. Alajarin and C. Burgosin Modern Heterocyclic Chemistry, ed. J. Alvarez-Builla, J. J. Vaquero, J. Barluenga, 2011, Vol. 3, pp 1527–1629 Search PubMed.
- J. Achan, A. O. Talisuna, A. Erhart, A. Yeka, J. K. Tibenderana, F. N. Baliraine, P. J. Rosenthal and U. D‘Alessandro, Malar. J., 2011, 10, 144 CrossRef CAS and references cited therein..
- (a) H. C. Richard, Tetrahedron, 1981, 37, 1047 CrossRef; (b) A. Perzyna, C. Marty, M. Facompré, J.-F. Goossens, N. Pommery, P. Colson, C. Houssier, R. Houssin, J.-P. Hénichart and C. Bailly, J. Med. Chem., 2002, 45, 5809 CrossRef CAS; (c) M. E. Wall, M. C. Wani, C. E. Cook, K. H. Palmer, A. T. McPhail and G. A. Sim, J. Am. Chem. Soc., 1966, 88, 3888 CrossRef CAS; (d) D. Wu, Tetrahedron, 2003, 59, 8649 CrossRef.
- (a) I.-S. Chen, I.-L. Tsai, S.-J. Wu, W.-S. Sheen, T. Ishikawa and H. Ishii, Phytochemistry, 1993, 34, 1449 CrossRef CAS; (b) D. C. Harrowven, M. I. T. Nunn, N. J. Blumire and D. R. Fenwick, Tetrahedron Lett., 2000, 41, 6681 CrossRef CAS; (c) D. C. Harrowven, M. I. T. Nunn, N. J. Blumire and D. R. Fenwick, Tetrahedron, 2001, 57, 4447 CrossRef CAS.
- (a) J. D. Maguire, E. Krisin, H. Marwoto, T. L. Richie, D. J. Fryauff and J. K. Baird, Clin. Infect. Dis., 2006, 42, 1067 CrossRef CAS; (b) J. Wiesner, R. Ortmann, H. Jomaa and M. Schlitzer, Angew. Chem., Int. Ed., 2003, 42, 5274 CrossRef CAS.
- (a) R. Engel in Handbook of Organophosphorus Chemistry, M. Dekker, Inc.New York, 1992 Search PubMed; (b) Fluorine in Medicinal Chemistry and Chemical Biology, ed. I. Ojima, John Wiley & Sons, 2009 Search PubMed; (c) V. D. Romanenko and V. P. Kukhar, Chem. Rev., 2006, 106, 3868 CrossRef CAS; (d) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef; (e) Fluorinated Heterocyclic Compounds: Synthesis, Chemistry and Applications, ed. V. A. Petrov, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009 Search PubMed; (f) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC; (g) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- R. Engel, Chem. Rev., 1977, 77, 349 CrossRef CAS.
- (a) C. C. Cheng and S. J. Yan, Org. React., John Wiley & Sons, New York, 1982, 28, 37 Search PubMed; (b) J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M. A. D. C. Carreiras and E. Soriano, Chem. Rev., 2009, 109, 2652 CrossRef CAS; (c) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Adv. Heterocycl. Chem., 2011, 102, 139 CrossRef CAS.
- F. W. Bergstrom, Chem. Rev., 1944, 35, 156 CrossRef.
- R. H. F. Manske and M. Kukla, Org. React., 1953, 7, 59 Search PubMed.
- F. W. Bergstrom, Chem. Rev., 1944, 35, 153 CrossRef.
- R. H. Reitsema, Chem. Rev., 1948, 43, 47 CrossRef.
- G. Jones in The Chemistry of Heterocyclic Compounds, ed. A. Weissberger and E. C. Taylor, John Wiley & Sons, Chichester, 1977, 32, Part I, pp 93–318 Search PubMed.
- (a) C. C. Cho, N. Y. Lee, T.-J. Kim and S. C. Shim, J. Heterocycl. Chem., 2004, 41, 409 CrossRef CAS; (b) A. Arcadi, M. Aschi, F. Marinelli and M. Verdecchia, Tetrahedron, 2008, 64, 5354 CrossRef CAS; (c) A. Arcadi, F. Marinelli and E. Rossi, Tetrahedron, 1999, 55, 13233 CrossRef CAS; (d) S. Cacchi, G. Fabrizi and F. Marinelli, Synlett, 1999, 401, 404 Search PubMed; (e) F. Nissen and H. Detert, Eur. J. Org. Chem., 2011, 2011, 2845 CrossRef; (f) R. Yanada, K. Hashimoto, R. Tokizane, Y. Miwa, H. Minami, K. Yanada, M. Ishikura and Y. Takemoto, J. Org. Chem., 2008, 73, 5135 CrossRef CAS; (g) X. Zhang, M. A. Campo, T. Yao and R. C. Larock, Org. Lett., 2005, 7, 763 CrossRef CAS; (h) X. Zhang, T. Yao, M. A. Campo and R. C. Larock, Tetrahedron, 2010, 66, 1177 CrossRef CAS; (i) J. Zhao, C. Peng, L. Liu, Y. Wang and Q. Zhu, J. Org. Chem., 2010, 75, 7502 CrossRef CAS.
- (a) H. Li, C. Wang, H. Huang, X. Xu and Y. Li, Tetrahedron Lett., 2011, 52, 1108 CrossRef CAS; (b) H. Li, X. Xu, J. Yang, X. Xie, H. Huang and Y. Li, Tetrahedron Lett., 2011, 52, 530 CrossRef CAS; (c) N. T. Patil and V. S. Raut, J. Org. Chem., 2010, 75, 6961 CrossRef CAS; (d) B. Madhav, S. N. Murthy, K. R. Rao and Y. V. D. Nageswar, Helv. Chim. Acta, 2010, 93, 257 CrossRef CAS; (e) J. B. Hendrickson, R. Rees and J. F. Templeton, J. Am. Chem. Soc., 1964, 86, 107 CrossRef CAS.
- (a) B. Duda, S. N. Tverdomed and G.-V. Röschenthaler, Org. Biomol. Chem., 2011, 9, 8228 RSC; (b) B. Duda, S. N. Tverdomed and G.-V. Röschenthaler, J. Org. Chem., 2011, 76, 71 CrossRef CAS.
- (a) P. Friedlander and S. Henriques, Chem. Ber., 1882, 15, 2572 CrossRef; (b) S. G. McGeachin, Can. J. Chem., 1966, 44, 2323 CrossRef CAS; (c) A. Abert and H. Yamamoto, J. Chem. Soc. B, 1966, 956 RSC.
- W. Borsche and W. Ried, Liebigs Ann. Chem., 1943, 554, 269 CrossRef CAS.
- H. K. Porter, Org. React., 1973, 20, 455 CAS.
- CCDC 884142 (for 4c) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viawww.ccdc.cam.ac.uk/data_request/cif.
- J. Vicente, M. T. Chicote, A. J. Martinez-Martinez, D. Bautista and P. G. Jones, Org. Biomol. Chem., 2011, 9, 2279 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and the copy of 1H, 13C, 19F, 31P NMR and HRMS spectra of new compounds. CCDC reference number 884142. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2ra21212a |
| This journal is © The Royal Society of Chemistry 2012 |