DOI:
10.1039/C2RA21343E
(Paper)
RSC Adv., 2012,
2, 9142-9150
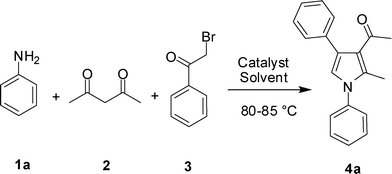
Yb(OTf)3 mediated MCR: a new and regioselective approach towards polysubstituted pyrroles of pharmacological interest†
Received
2nd July 2012
, Accepted 7th August 2012
First published on 8th August 2012
Abstract
A regioselective synthesis of 1,2,3,4-tetrasubstituted pyrroles has been achieved via Yb(OTf)3-mediated 3-component reaction of amines, a 1,3-diketone and phenacyl bromide in a single pot. Yb(OTf)3 was identified as a reusable catalyst and a number of pyrrole derivatives were prepared by using this strategy. Single crystal X-ray diffraction study of a representative compound confirmed the substitution pattern on the central pyrrole ring. The crystal structure analysis of the same compound indicated the presence of a sheet-like molecular arrangement along the bc-plane present in the molecule. A possible mechanism for the regioselective formation of 1,2,3,4-tetrasubstituted pyrrole rings is discussed. A number of compounds synthesized showed PDE4 inhibitory properties when tested in vitro and two of them were identified as inhibitors of further interest.
Introduction
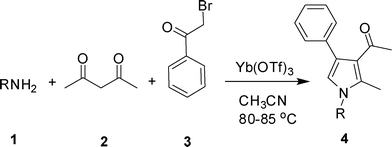
Besides their occurrences in nature1 (e.g. heme, chlorophyll, vitamin B12 etc), pyrroles have found wide applications in material science,2 organic synthesis,3 medicinal chemistry and drug discovery.4 The anticancer drug candidate tallimustine and blockbuster lipid lowering drug atorvastatin belong to this class of N-heterocycles. While a number of classic methods including Knorr,5 Paal–Knorr,6 and Hantzsch syntheses7 are known for the synthesis of pyrrole derivatives, many of them involve tedious multistep synthetic operations. Recently, strategies based on palladium-, platinum-, copper- and gold-catalysed cycloisomerisation or cyclisation reactions of a variety of acyclic precursors, such as alkynylimines,8a,b (Z)-(2-en-4-ynyl)amines,8c homopropargyl azides,8d,e or other precursors8f,g have been reported. In spite of these outstanding efforts, it is still challenging to prepare polysubstituted pyrroles possessing diverse substituents directly from readily available building blocks in a single synthetic operation. Multi-component domino reactions (MCRs) are known to provide structurally complex molecules in a single pot ensuring high atom economy and good overall yields.9 Thus, synthesis of functionalized pyrroles has been reported via MCR catalyzed by Fe(III),10 Pd11a and I2.11b However, a search for another environmentally friendly catalyst is still highly desirable. As mild and selective catalysts, lanthanide triflates have been used widely in C–C and C–X bond-forming reactions.12 Unlike classical Lewis acids that are often used in stoichiometric quantities, lanthanide triflates effectively promote these reactions in catalytic quantities. Additionally, lanthanide triflates are water tolerant and can be recovered and reused without loss of activity. The use of Yb(OTf)3 as an efficient catalyst in MCRs or similar reactions has been explored earlier.13 For example, synthesis of 3,4,5-trisubstituted-3,6-dihydro-2H-1,3-oxazines13a or polyhydroquinoline derivatives13c has been carried out using Yb(OTf)3-promoted one-pot three or four component reactions, respectively. In continuation of our efforts on the development of new MCRs for the synthesis of various heterocyclic structures,14 we now wish to report a direct route to 1,2,3,4-tetrasubstituted pyrroles via Yb(OTf)3-catalyzed three component reaction of amines (1), a 1,3-diketone (2) and phenacyl bromide (3) (Scheme 1). To the best of our knowledge, use of this strategy for the synthesis of 1,2,3,4-functionalized pyrroles is not known in the literature.
 |
| | Scheme 1 Yb(OTf)3-mediated synthesis of 1,2,3,4-tetrasubstituted pyrroles. | |
Results and discussion
Initially, we examined the reaction of aniline (1), acetyl acetone (2) and phenacyl bromide (3) in CH3CN in the presence of several catalysts e.g. p-toluenesulfonic acid (p-TSA), amberlyst-15, iodine and NaHSO3 (2.5%) absorbed on silica (entries 1–4, Table 1). While the regioselective formation of 1-(2-methyl-1,4-diphenyl-1H-pyrrol-3-yl)ethanone (4a) was observed, in these cases the product was isolated in poor or negligible yield. The use of Yb(OTf)3 as a catalyst however was found to be beneficial and the reaction was completed within 8 h affording the product 4a in 75% yield (entry 5, Table 1). To test the recyclability of the catalyst, Yb(OTf)3 was recovered by simple filtration and reused, whereupon 4a was isolated without significant decrease in yield. The yield of 4a was found to be 72, 65 and 60% after 1st, 2nd and 3rd recovery and reuse of the catalyst. An increase of the reaction time did not improve the product yield. The use of other solvents such as DMF, THF and toluene (entries 6–8, Table 1) were examined but found to be less effective in terms of product yield. While all these reactions were carried out at 80–85 °C, the MCR became less efficient when conducted at room temperature (entry 9, Table 1. Overall, the use of Yb(OTf)3 in acetonitrile at 80–85 °C was identified as the optimum conditions for the present MCR.
Table 1 Effect of reaction conditions on the MCR of 1a, 2 and 3a
| Entry |
Catalyst (mol%)b |
Solvent |
Time (h) |
Yield (%)c |
|
All the reactions were carried out using 1a (1.5 mmol), 2 (1.0 mmol), 3 (1.0 mmol) at 80–85 °C.
With respect to the aniline 1a used.
Isolated yield.
Recovered Yb(OTf)3 was used.
The reaction was carried out at room temperature.
|
| 1 |
p-TSA (2.5%) |
CH3CN |
24 |
10 |
| 2 |
Amberlyst-15 (2.5%) |
CH3CN |
24 |
7 |
| 3 |
Iodine (2.5%) |
CH3CN |
24 |
16 |
| 4 |
NaHSO3 (2.5%) absorbed on silica |
CH3CN |
24 |
11 |
| 5 |
Yb(OTf)3 (0.1%) |
CH3CN |
8 |
75 (72)d |
| 6 |
Yb(OTf)3 (0.1%) |
DMF |
8 |
40 |
| 7 |
Yb(OTf)3 (0.1%) |
THF |
8 |
20 |
| 8 |
Yb(OTf)3 (0.1%) |
Toluene |
8 |
30 |
| 9 |
Yb(OTf)3 (0.1%) |
CH3CN |
24e |
10 |
Having the optimized reaction conditions in hand, we then examined the further scope of this methodology. Thus, a variety of 1,2,3,4-tetrasubstituted pyrroles (4) were synthesized in good yields (Table 2). Various amines such as aryl, alkylaryl and alkyl amines were employed in the present MCR, all of which participated well. Substituents like Cl, Br, OMe, OH and NO2 present in the amines were well tolerated (Table 2). We then examined the use of 2-bromo-1-(4-methoxyphenyl)ethanone (3a) in place of phenacyl bromide (3) in the present MCR, the other two reactants being 1g and 2. The desired product 1-[1,4-bis(4-methoxyphenyl)-2-methyl-1H-pyrrol-3-yl]-ethanone (5) was isolated in 63% yield after 7.5 h. All the compounds synthesized were characterized by spectral (1H and 13C NMR, IR and HRMS) data and this was supported by the molecular structure of 4m being confirmed unambiguously by X-ray analysis (Fig. 1).15 This also confirmed the 1,2,3,4-substitution pattern on the central pyrrole ring of this class of molecules. Compound 4m crystallized in the monoclinic P21/c space group with one molecule in the asymmetric unit (Z = 4, Z′ = 1) (Fig. 1). The molecule in the asymmetric unit has no conventional functional groups to form strong hydrogen bonds. The molecule contains a keto and a chloro group that are responsible for the formation of weak Van der Waals interactions. The molecule in the asymmetric unit forms a channel-like tetramer through C–O⋯H and C–Cl⋯N interactions, which leads to the formation of ladder-type packing as shown in Fig. 2. These interactions propagated in 2D sheets along the bc-plane as shown in Fig. 3.
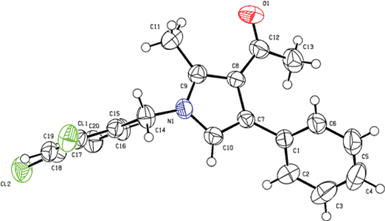 |
| | Fig. 1 ORTEP representation of 4m. (Thermal ellipsoids are drawn at 50% probability level.) | |
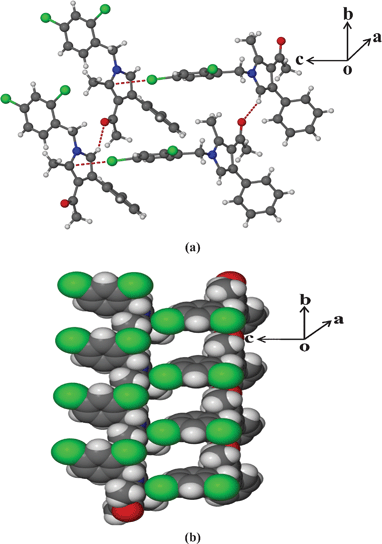 |
| | Fig. 2 (a) Tetramer formation via C–O⋯H and C–Cl⋯N interactions and (b) ladder formation in 4m. | |
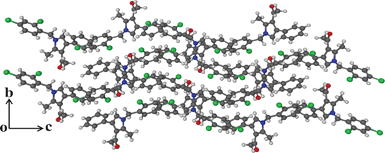 |
| | Fig. 3 Sheet-like molecular arrangement along the bc-plane present in the molecule 4m. | |
| Entry |
Amines (1) |
Products (4)b |
Time (h) |
Yield (%)c |
|
All the reactions were carried out using amine (1) (1.5 mmol), acetyl acetone (2) (1.0 mmol), phenacyl bromide (3) (1.0 mmol) at 80–85 °C.
Identified by 1H NMR, IR and MS.
Isolated yields.
|
| 1 |
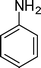 1a
1a
|
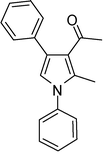 4a
4a
|
9 |
75 |
| 2 |
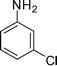 1b
1b
|
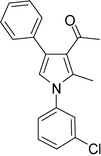 4b
4b
|
7 |
55 |
| 3 |
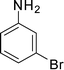 1c
1c
|
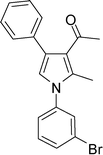 4c
4c
|
7 |
55 |
| 4 |
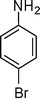 1d
1d
|
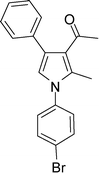 4d
4d
|
5 |
65 |
| 5 |
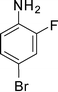 1e
1e
|
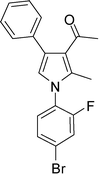 4e
4e
|
10 |
65 |
| 6 |
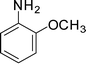 1f
1f
|
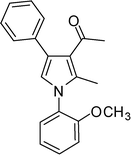 4f
4f
|
8 |
60 |
| 7 |
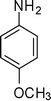 1g
1g
|
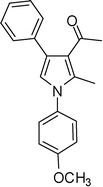 4g
4g
|
7.5 |
69 |
| 8 |
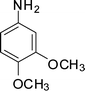 1h
1h
|
 4h
4h
|
7 |
60 |
| 9 |
 1i
1i
|
 4i
4i
|
7 |
72 |
| 10 |
 1j
1j
|
 4j
4j
|
8 |
75 |
| 11 |
 1k
1k
|
 4k
4k
|
8 |
55 |
| 12 |
 1l
1l
|
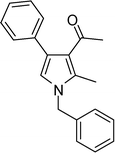 4l
4l
|
7 |
72 |
| 13 |
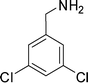 1m
1m
|
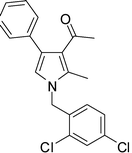 4m
4m
|
12 |
75 |
| 14 |
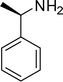 1n
1n
|
 4n
4n
|
10 |
60 |
| 15 |
 1o
1o
|
 4o
4o
|
10 |
70 |
We have shown that the present Yb(OTf)3-mediated three component reaction of amines (1), acetyl acetone (2) and phenacyl bromide (3) afforded 1,2,3,4-tetrasubstituted pyrroles. The MCR of 1a, 2 and 3 was also carried out under the conditions of entry 5 of Table 1 using Sc(OTf)3 in place of Yb(OTf)3. The reaction proceeded to give the desired product 4a but in low yield indicating the superior catalytic activity of Yb(OTf)3 in the present MCR. The use of other lanthanide triflates however has not yet been tested but could form the basis of future studies.
Mechanistically, the reaction seems to proceed via the usual Hantzsch mechanism, in which a β-enaminone (E-1) is first formed by reaction of the β-dicarbonyl (2) and amine reactants (1) (Scheme 2).16 It is expected that Yb(OTf)3 coordinates with the carbonyl oxygen of 2 thereby activating it for a nucleophilic attack. A subsequent reaction between E-1 and the α-bromo ketone (3) (activated by Yb(OTf)3) provides the intermediate E-2 which on intramolecular cyclization followed by dehydration provides pyrrole 4. To gain further evidence on the intermediacy of E-1, the β-enaminone obtained via Yb(OTf)3-mediated reaction of aniline (1) and acetyl acetone (2) was reacted with phenacyl bromide (3) in the presence of Yb(OTf)3 in CH3CN at 80–85 °C. The reaction was completed within 4 h providing the desired compound 4a in 70% yield indicating the intermediacy of E-1 in the present MCR. It is worth mentioning that a similar MCR in water in the presence of β-cyclodextrin as a supramolecular catalyst provided regioisomeric 1,2,3,5-tetrasubstituted pyrroles in good yields.17 The role of cyclodextrin was thought to be in the activation of phenacyl bromide through H–bonding interactions of its bromine atom with OH substituents in the cyclodextrin rim. The reaction therefore proceeded via a path other than the usual Hantzsch mechanism, providing the regioisomeric pyrroles.
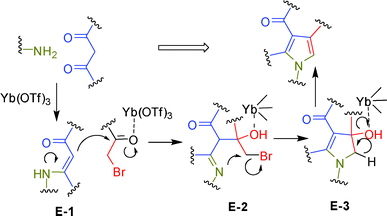 |
| | Scheme 2 Proposed mechanism for the Yb(OTf)3-mediated synthesis of pyrrole. | |
Due to our interest in pyrrole based inhibitors of PDE4,11a some of the compounds synthesized were tested for their PDE4B inhibitory properties in vitro. Inhibitors of PDE4 are known to be beneficial for the treatment of asthma and chronic obstructive pulmonary disease (COPD).18 However, clinical studies of the PDE4 inhibitor rolipram were limited by side effects including nausea and emesis that were thought to arise from inhibition of the PDE4D subtype.19 Similarly, side effects have restricted the therapeutic index of the second-generation PDE4 inhibitors cilomilast and roflumilast.20 Selective inhibition of the PDE4B subtype may provide a means to achieve efficacy while potentially mitigating these adverse events.21 The compounds were tested at 30 μM using a PDE4B enzyme assay22 (Table 3). Rolipram23 was used as a reference compound in this assay. Compounds 4g and 4n showed significant inhibition of PDE4B (Table 3) suggesting that the present class of pyrroles may have medicinal value. In a dose–response study, compound 4n showed dose dependent inhibition i.e. 20, 40, 61, 69 and 75% inhibition of PDE4B when tested at concentrations of 1, 5, 10, 30 and 100 μM.
Table 3 Inhibition of PDE4B by compound 4 at 30 μM
| Entry |
Compounds |
Average % inhibition |
SDa |
|
SD = standard deviation.
|
| 1 |
4d
|
37.04 |
0.75 |
| 2 |
4e
|
28.59 |
5.36 |
| 3 |
4g
|
57.89 |
2.31 |
| 4 |
4j
|
37.84 |
0.04 |
| 5 |
4k
|
25.91 |
6.00 |
| 6 |
4n
|
66.13 |
1.18 |
| 7 |
4m
|
46.76 |
0.86 |
| 8 |
4d
|
37.04 |
0.75 |
In order to understand the nature of the interactions of these molecules with PDE4B, docking studies were carried out using the compounds 4g and 4n. Glide scores of −5.26 and −7.04 kcal mol−1 were obtained for 4g and 4n respectively. The interaction of compound 4g with the PDE4B protein (Fig. 4, see ESI† also) mainly consisted of π–π stacking interactions between (i) the benzene ring of pyrrole 4g and histidine (His234) and (ii) the pyrrole ring and histidine (His234). Additionally, the acetyl group of 4g was involved in ionic interactions with Mg and Zn ions. Similarly, the interaction of compound 4n with the PDE4B protein (Fig. 5, ESI†) consisted of a π–π stacking interaction between the benzene ring of 4n and histidine (His234), and a hydrogen bond between the carbonyl oxygen of pyrrole and glutamine 443.
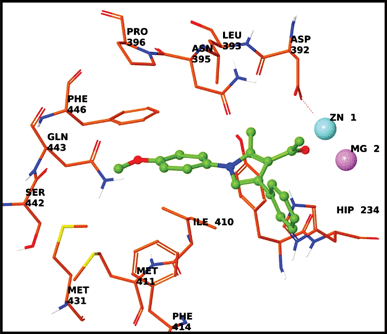 |
| | Fig. 4 Docking of 4g in the active site of PDE4B. | |
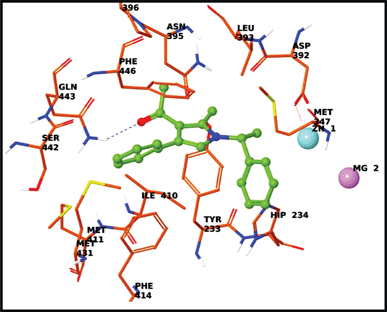 |
| | Fig. 5 Docking of 4n in the active site of PDE4B. | |
Conclusions
In conclusion, a direct and regioselective synthesis of polysubstituted pyrroles has been achieved via Yb(OTf)3-mediated 3-component reaction in a single pot. A MCR that involved the reaction of amines, a 1,3-diketone and phenacyl bromide was used to prepare a number of pyrrole derivatives in good yields. The catalyst Yb(OTf)3 can be recovered and reused without significant loss of activity. Single crystal X-ray diffraction study of a representative compound confirmed the 1,2,3,4-substitution pattern on the central pyrrole ring. The crystal structure analysis of the same compound indicated the presence of a sheet-like molecular arrangement along the bc-plane present in the molecule. A possible mechanism and rationale for the regioselective formation of 1,2,3,4-tetrasubstituted pyrrole rings is presented. Some of the compounds synthesized showed PDE4 inhibitory properties when tested in vitro and two of them were found to be promising. Overall, this research demonstrates a greener approach in the medicinal chemistry effort towards the identification of new PDE4 inhibitors. The present Yb(OTf)3-mediated MCR would therefore find usage in building libraries of small molecules based on the 1,2,3,4-tetrasubstituted pyrrole framework of potential pharmacological interest.
Experimental section
General methods
Unless stated otherwise, reactions were performed under nitrogen atmosphere using oven dried glassware. Reactions were monitored by thin layer chromatography (TLC) on silica gel plates (60 F254), visualizing with ultraviolet light or iodine spray. Flash chromatography was performed on silica gel (230–400 mesh) using distilled hexane, ethyl acetate, dichloromethane. 1H NMR and 13C NMR spectra were recorded in CDCl3 solution by using 400 and 100 MHz spectrometers, respectively. Proton chemical shifts (δ) are relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in ppm. Spin multiplicities are given as s (singlet), d (doublet), t (triplet) and m (multiplet) as well as b (broad). Coupling constants (J) are given in hertz. Infrared spectra were recorded on a FT-IR spectrometer. Melting points (mp) were determined using a Buchi B-540 melting point apparatus and are uncorrected. MS spectra were obtained on an Agilent 6430 series Triple Quard LC-MS/MS spectrometer. High-resolution mass spectra (HRMS) were recorded using a Waters LCT Premier XE instrument.
General procedure for the preparation of 4
A mixture of amine 1 (1.5 mmol), acetyl acetone 2 (1.0 mmol), phenacyl bromide 3 (1.0 mmol) and Yb(OTf)3 (0.1 mol%) in acetonitrile (5 mL) was stirred for 30 min at 30–35 °C and then heated at 80–85 °C for 5–10 h. After completion of the reaction, the mixture was cooled to room temperature and concentrated under vacuum. The residue was purified by silica gel column using ethyl acetate–hexanes to give the desired product.
1-(2-Methyl-1,4-diphenyl-1H-pyrrol-3-yl)ethanone (4a)
Brown sticky liquid; IR (neat) 2885, 1649, 1598, 1498, 1407, 1215, 1032 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.07 (s, 3H), 2.41 (s, 3H), 6.67 (s, 1H), 7.30–7.51 (m, 10H); 13C NMR (CDCl3, 100 MHz) δ 12.9, 29.7, 120.6 (2C), 126.2 (3C), 126.8, 128.1, 128.3 (2C), 129.3 (4C), 135.3, 136.0, 138.7, 197.7; HRMS: m/z calcd for C19H18NO (M + 1) 276.1388; found 276.1407.
1-(1-(3-Chlorophenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4b)
Brown sticky liquid; IR (neat) 2928, 1947, 1651, 1594, 1402, 1276, 1008, 668 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.06 (s, 3H), 2.41 (s, 3H), 6.65 (s, 1H), 7.23–7.25 (m, 2H), 7.31–7.43 (m, 7H); 13C NMR (CDCl3, 100 MHz) δ 12.8, 31.1, 120.3, 122.9, 124.4, 126.5, 126.6, 126.9, 128.3 (3C), 129.3 (2C), 130.3, 135.0, 135.1, 135.7, 139.8, 197.6; HRMS: m/z calcd for C19H17ClNO (M + 1) 310.0999; found 310.1001.
1-(1-(3-Bromophenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4c)
Brown sticky liquid; IR (neat), 2981, 1653, 1603, 1495, 1411, 1215, 755 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.06 (s, 3H), 2.41 (s, 3H), 6.65 (s, 1H), 7.27–7.40 (m, 7H), 7.52–7.58 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 12.8, 31.1, 120.3, 122.8, 122.9, 124.9, 126.6, 126.9, 128.3 (2C), 129.3 (2C), 129.4, 130.6, 131.2, 135.1, 135.7, 139.9, 197.6; HRMS: m/z calcd for C19H17BrNO (M + 1) 354.0494; found 354.0484.
1-(1-(4-Bromophenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4d)
Light brown solid; mp 138–139 °C; IR (neat) 2981, 1652, 1603, 1410, 1215, 756 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.06 (s, 3H), 2.39 (s, 3H), 6.63 (s, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.31–7.38 (m, 5H), 7.63 (dd, J = 8.0 and 2.0 Hz, 2H); 13C NMR (CDCl3, 100 MHz) δ 12.8, 31.1, 120.3, 122.0, 122.9, 126.6, 126.9, 127.8 (2C), 128.3 (2C), 129.3 (2C), 132.6 (2C), 135.0, 135.7, 137.7, 197.6; HRMS: m/z calcd for C19H17BrNO (M + 1) 354.0494; found 354.0479.
1-(1-(4-Bromo-2-fluorophenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4e)
Brown solid; mp 100–101 °C: IR (neat) 2982, 1652, 1603, 1410, 1215, 755 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.06 (s, 3H), 2.33 (s, 3H), 6.58 (s, 1H), 7.23–7.26 (m, 1H), 7.37–7.52 (m, 7H); 13C NMR (CDCl3, 100 MHz) δ 12.2, 31.0, 120.4, 120.7, 122.7, 122.8, 125.6, 125.8, 126.8, 127.0 (2C), 128.2, 129.1, 129.3, 130.0, 135.8, 158.3, 197.5; HRMS: m/z calcd for C19H16BrFNO (M + 1) 372.0384; found 372.0399.
1-(1-(2-Methoxyphenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4f)
Brown sticky liquid; IR (neat) 2962, 1885, 1651, 1547, 1444, 1250, 1035 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.08 (s, 3H), 2.28 (s, 3H), 3.83 (s, 3H), 6.56 (s, 1H), 7.02–7.08 (m, 2H), 7.23–7.31 (m, 3H), 7.32–7.43 (m, 4H); 13C NMR (CDCl3, 100 MHz) δ 12.4, 31.1, 55.7, 112.0, 120.7, 121.0, 121.6, 125.9, 126.6, 127.4, 127.7, 127.9, 128.1, 128.8, 129.4, 129.9, 136.3, 137.1, 154.7, 197.5; HRMS: m/z calcd for C20H20NO2 (M + 1) 306.1494; found 306.1498.
1-(1-(4-Methoxyphenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4g)
White solid; mp 87.5–88.5 °C; IR (neat) 2936, 1884, 1650, 1515, 1408, 1216, 1034 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.07 (s, 3H), 2.37 (s, 3H), 3.86 (s, 3H), 6.62 (s, 1H), 6.99 (dd, J = 8.0 and 2.0 Hz, 2H), 7.22–7.29 (m, 2H), 7.31–7.38 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 12.8, 31.0, 55.5, 114.4 (2C), 120.8, 122.2, 126.0, 126.7, 127.4 (2C), 128.2 (2C), 129.3 (2C), 131.6, 135.6, 136.1, 159.2, 197.6; HRMS: m/z calcd for C20H20NO2 (M + 1) 306.1494; found 306.1498.
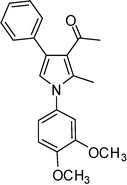
1-(1-(3,4-Dimethoxyphenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4h)
White solid; mp 127–128 °C; IR (neat) 2824, 1802, 1652, 1588, 1421, 1215, 1072 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.07 (s, 3H), 2.39 (s, 3H), 3.89 (s, 3H), 3.94 (s, 3H), 6.65 (s, 1H), 6.82–6.95 (m, 3H), 7.31–7.38 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 12.8, 31.0, 56.1 (2C), 109.9, 111.0, 118.5, 120.8, 122.2, 126.0, 126.8, 128.2 (2C), 129.3 (2C), 131.7, 135.6, 136.0, 148.9, 149.2, 197.6; HRMS: m/z calcd for C21H22NO3 (M + 1) 336.1600; found 336.1584.
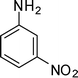
1-(2-Methyl-1-(3-nitrophenyl)-4-phenyl-1H-pyrrol-3-yl)ethanone (4i)
Pale yellow solid; mp 115–116 °C; IR (neat) 2981, 1802, 1654, 1535, 1404, 1215, 928, 669 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.07 (s, 3H), 2.44 (s, 3H), 6.72 (s, 1H), 7.34–7.40 (m, 5H), 7.70–7.75 (m, 2H), 8.25–8.31 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 12.9, 31.1, 120.1, 121.2, 122.8, 123.5, 127.2 (2C), 128.4 (2C), 129.2 (2C), 130.4, 131.9, 134.8, 135.3, 139.8, 148.7, 197.6; HRMS: m/z calcd for C19H17N2O3 (M + 1) 321.1239; found 321.1233.
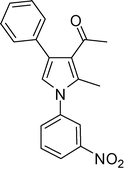
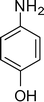
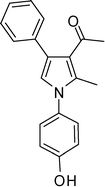
1-(1-(4-Hydroxyphenyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4j)
Off white solid; mp 143–144 °C; IR (neat) 1702, 1632, 1552, 1412, 1282, 1071 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.09 (s, 3H), 2.38 (s, 3H), 6.62 (s, 1H), 6.96 (dd, J = 2.0 Hz, 2H), 7.18 (dd, J = 2.0 Hz, 2H), 7.36–7.38 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 13.0, 30.1, 116.1 (2C), 121.2, 121.8, 126.2, 126.8, 127.5 (2C), 128.3 (2C), 129.3 (2C), 131.0, 136.0, 136.4, 156.3, 198.6; HRMS: m/z calcd for C19H18NO2 (M + 1) 292.1338; found 292.1365.
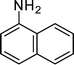
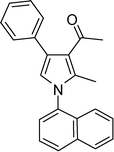
1-(2-Methyl-1-(naphthalen-1-yl)-4-phenyl-1H-pyrrol-3-yl)ethanone (4k)
Dark brown sticky liquid; IR (neat) 2933, 1922, 1601, 1573, 1514, 1434, 1313, 1281 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.14 (s, 3H), 2.22 (s, 3H), 6.71 (s, 1H), 7.30–7.58 (m, 10H), 7.94–7.98 (m, 2H); 13C NMR (CDCl3, 100 MHz) δ 12.4, 31.1, 121.7, 121.8, 122.9, 125.2, 125.3, 126.1, 126.8, 126.9 (2C), 127.6 (2C), 128.2, 128.3, 129.3, 129.4, 130.6, 134.1, 135.3, 136.1, 137.1, 197.7; HRMS: m/z calcd for C23H20NO (M + 1) 326.1545; found 326.1537.
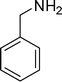
1-(1-Benzyl-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4l)
Brown solid; mp 52–54 °C; IR (neat) 2929, 1887, 1647, 1560, 1453, 1354, 1215, 1073 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.03 (s, 3H), 2.43 (s, 3H), 5.05 (s, 2H), 6.53 (s, 1H), 7.09 (d, J = 6.8 Hz, 2H), 7.25–7.55 (m, 8H); 13C NMR (CDCl3, 100 MHz) δ 11.5, 31.0, 50.2, 120.0 (2C), 122.0, 125.8, 126.6 (2C), 127.7, 128.1 (2C), 128.8 (2C), 129.3 (2C), 135.1, 136.2, 136.5, 197.5; HRMS: m/z calcd for C20H20NO (M + 1) 290.1545; found 290.1572.
1-(1-(2,4-Dichlorobenzyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4m)
White solid; mp 162–163 °C; IR (neat) 2927, 1949, 1652, 1549, 1507, 1402, 1215, 951, 669 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.04 (s, 3H), 2.40 (s, 3H), 5.09 (s, 2H), 6.50 (s, 1H), 6.60 (d, J = 8.4 Hz, 1H), 7.18 (d, J = 2.0 Hz, 1H), 7.28–7.39 (m, 5H), 7.43 (d, J = 2.0 Hz, 1H); 13C NMR (CDCl3, 100 MHz) δ 11.3, 31.1, 47.5, 119.9, 122.3, 126.4, 126.8, 127.8, 128.3, 128.6 (2C), 129.3 (2C), 129.4, 132.8, 133.0, 134.3, 135.0, 136.0, 197.6; HRMS: m/z calcd for C20H18Cl2NO (M + 1) 358.0765; found 358.0762.
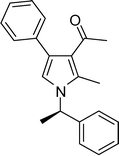
1-(2-Methyl-4-phenyl-1-((R)-1-phenylethyl)-1H-pyrrol-3-yl)ethanone (4n)
White solid; mp 127–128 °C; IR (neat) 2928, 1883, 1650, 1515, 1465, 1326, 1215, 1035 cm−1; 1H NMR (CDCl3, 400 MHz) δ 1.83 (d, J = 6.8 Hz, 3H), 2.03 (s, 3H), 2.41 (s, 3H), 5.36 (d, J = 6.8 Hz, 1H), 6.67 (s, 1H), 7.10 (d, J = 7.6 Hz, 2H), 7.25–7.37 (m, 8H); 13C NMR (CDCl3, 100 MHz) δ 11.5, 22.1, 31.1, 54.9, 116.5, 121.9, 125.6, 125.9, 126.6, 127.6, 127.7, 128.2, 128.3 (2C), 128.8, 129.3, 132.6, 135.0, 136.5, 141.9, 197.8; HRMS: m/z calcd for C21H22NO (M + 1) 304.1701; found 304.1718.
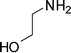
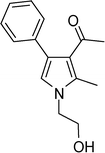
1-(1-(2-Hydroxyethyl)-2-methyl-4-phenyl-1H-pyrrol-3-yl)ethanone (4o)
Dark brown sticky liquid; IR (neat) 2924, 1702, 1632, 1551, 1413, 1282, 1070 cm−1; 1H NMR (CDCl3, 400 MHz) δ 2.00 (s, 3H), 2.49 (s, 3H), 3.89 (t, J = 5.2 Hz, 2H), 4.00 (t, J = 5.2 Hz, 2H), 6.57 (s, 1H), 7.26–7.40 (m, 5H); 13C NMR (CDCl3, 100 MHz) δ 11.5, 30.9, 48.5, 61.9, 119.9, 121.8, 125.9, 126.6, 128.2 (2C), 129.2 (2C), 135.1, 136.1, 197.8; HRMS: m/z calcd for C15H18NO2 (M + 1) 244.1338; found 244.1349.
1-[1,4-Bis(4-methoxyphenyl)-2-methyl-1H-pyrrol-3-yl]-ethanone (5)
A mixture of 4-methoxyaniline 1g (1.5 mmol), acetyl acetone 2 (1.0 mmol), 2-bromo-1-(4-methoxyphenyl)ethanone 3a (1.0 mmol) and Yb(OTf)3 (0.1 mol%) in acetonitrile (5 mL) was stirred for 30 min at 30–35 °C and then heated at 80–85 °C for 7.5 h. After completion of the reaction, the mixture was cooled to room temperature and concentrated under vacuum. The residue was purified by silica gel column using ethyl acetate–hexanes to give the desired product 5 as a light brown solid; mp 130–132 °C (lit10 133–135 °C).
Single crystal X-ray data for compound 4m
Single crystals suitable for X-ray diffraction of 4m were grown from methanol. The crystals were carefully chosen using a stereo zoom microscope supported by a rotatable polarizing stage. The data were collected at room temperature on Bruker's KAPPA APEX II CCD Duo with graphite monochromatic Mo-Kα radiation (0.71073 Å). The crystals were glued to a thin glass fibre using FOMBLIN immersion oil and mounted on the diffractometer. The intensity data were processed using Bruker's suite of data processing programs (SAINT), and absorption corrections were applied using SADABS.24 The crystal structure was solved by direct methods using SHELXS-97 and the data was refined by full matrix least-squares refinement on F2 with anisotropic displacement parameters for non-H atoms, using SHELXL-97.25
Crystal data of 4m: molecular formula = C20H17Cl2NO, formula weight = 357.07, crystal system = monoclinic, space group = P21/c, a = 12.536(2) Å, b = 7.767(11) Å, c = 18.601(3) Å, V = 1756.2(5) Å3, T = 296(2) K, Z = 4, Dc = 1.355 mg m−3, μ(Mo-Kα) = 0.38 mm−1, 18148 reflections measured, 4357 independent reflections, 2443 observed reflections [I > 2.0 σ (I)], R1_obs = 0.064, goodness of fit =1.01. Crystallographic data (excluding structure factors) for 4m have been deposited with the Cambridge Crystallographic Data Center as supplementary publication number CCDC 867480.†
Pharmacology
Materials and methods
Cells and reagents: HEK 293 and Sf9 cells were obtained from ATCC (Washington D.C., USA). HEK 293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (Invitrogen Inc., San Diego, CA, USA). Sf9 cells were routinely maintained in Grace's supplemented medium (Invitrogen) with 10% FBS. RAW 264.7 cells (murine macrophage cell line) were obtained from ATCC and routinely cultured in RPMI 1640 medium with 10% fetal bovine serum (Invitrogen Inc.). cAMP was purchased from SISCO Research Laboratories (Mumbai, India). PDElight HTS cAMP phosphodiesterase assay kit was procured from Lonza (Basel, Switzerland). PDE4B1 clone was from OriGene Technologies (Rockville, MD, USA).
PDE4B cDNA was sub-cloned into pFAST Bac HTB vector (Invitrogen) and transformed into DH10Bac (Invitrogen) competent cells. Recombinant bacmids were tested for integration by PCR analysis. Sf9 cells were transfected with bacmid using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Subsequently, P3 viral titer was amplified, cells were infected and 48 h post infection cells were lysed in lysis buffer (50 mM Tris-HCl pH 8.5, 10 mM 2-mercaptoethanol, 1% protease inhibitor cocktail (Roche), 1% NP40). Recombinant His-tagged PDE4B protein was purified as previously described elsewhere.22 Briefly, the lysate was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min at 4 °C and the supernatant was collected. The supernatant was mixed with Ni-NTA resin (GE Life Sciences) in a ratio of 4:1 (v/v) and equilibrated with binding buffer (20 mM Tris-HCl pH 8.0, 500 mM KCl, 5 mM imidazole, 10 mM 2-mercaptoethanol and 10% glycerol) in a ratio of 2:1 (v/v) and mixed gently on rotary shaker for 1 h at 4 °C. After incubation, the lysate–Ni-NTA mixture was centrifuged at 4500 rpm for 5 min at 4 °C and the supernatant was collected as the flow-through fraction. The resin was washed twice with wash buffer (20 mM Tris-HCl pH 8.5, 1 M KCl, 10 mM 2-mercaptoethanol and 10% glycerol). The protein was eluted sequentially twice using elution buffers (Buffer I: 20 mM Tris-HCl pH 8.5, 100 mM KCl, 250 mM imidazole, 10 mM 2-mercaptoethanol, 10% glycerol, Buffer II: 20 mM Tris-HCl pH 8.5, 100 mM KCl, 500 mM imidazole, 10 mM 2-mercaptoethanol, 10% glycerol). Eluates were collected in four fractions and analyzed by SDS-PAGE. Eluates containing PDE4B protein were pooled and stored at −80 °C in 50% glycerol until further use.
000 rpm for 10 min at 4 °C and the supernatant was collected. The supernatant was mixed with Ni-NTA resin (GE Life Sciences) in a ratio of 4:1 (v/v) and equilibrated with binding buffer (20 mM Tris-HCl pH 8.0, 500 mM KCl, 5 mM imidazole, 10 mM 2-mercaptoethanol and 10% glycerol) in a ratio of 2:1 (v/v) and mixed gently on rotary shaker for 1 h at 4 °C. After incubation, the lysate–Ni-NTA mixture was centrifuged at 4500 rpm for 5 min at 4 °C and the supernatant was collected as the flow-through fraction. The resin was washed twice with wash buffer (20 mM Tris-HCl pH 8.5, 1 M KCl, 10 mM 2-mercaptoethanol and 10% glycerol). The protein was eluted sequentially twice using elution buffers (Buffer I: 20 mM Tris-HCl pH 8.5, 100 mM KCl, 250 mM imidazole, 10 mM 2-mercaptoethanol, 10% glycerol, Buffer II: 20 mM Tris-HCl pH 8.5, 100 mM KCl, 500 mM imidazole, 10 mM 2-mercaptoethanol, 10% glycerol). Eluates were collected in four fractions and analyzed by SDS-PAGE. Eluates containing PDE4B protein were pooled and stored at −80 °C in 50% glycerol until further use.
PDE4B enzymatic assay
The inhibition of PDE4B enzyme was measured using PDElight HTS cAMP phosphodiesterase assay kit (Lonza) according to the manufacturer's recommendations. Briefly, 10 ng of PDE4B enzyme was pre-incubated either with DMSO (vehicle control) or compound for 15 min before incubation with the substrate cAMP (5 μM) for 1 h. The reaction was halted with a stop solution followed by incubation with a detection reagent for 10 min in the dark. Luminescence values (RLUs) were measured by a Multilabel plate reader (Perklin Elmer 1420 Multilabel counter). The percentage of inhibition was calculated using the following formula:| | 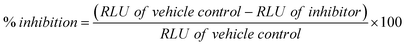 |
(1)
|
Acknowledgements
GRR thanks Dr V. Dahanukar and Dr U. K. Syam Kumar for encouragement and the analytical group of DRL for spectral support. MP thanks Prof J. Iqbal for support.
References
-
(a)
R. J. Sundburg, in Comprehensive Heterocyclic Chemistry II; A. R. Katritzky, C. W. Rees and E. F. V. Scriven, ed.; Pergamon Press: Oxford, 1996; Vol. 2, p. 119 Search PubMed;
(b) H. Fan, J. Peng, M. T. Hamann and J. -F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS , and references cited here.
-
(a) For a review, see: S. J. Higgins, Chem. Soc. Rev., 1997, 26, 247 RSC;
(b)
L. Groenendaal, E.-W. Meijer and J. A. J. M. Vekemans, in Electronic Materials: The Oligomer Approach; K. Müllen and G. Wegner, ed.; Wiley-VCH: Weinheim, 1997 Search PubMed;
(c) P. Novak, K. Muller, S. V. Santhanam and O. Hass, Chem. Rev., 1997, 97, 207 CrossRef CAS;
(d) S. Gabriel, M. Cecius, K. Fleury-Frenette, D. Cossement, M. Hecq, N. Ruth, R. Jerome and C. Jerome, Chem. Mater., 2007, 19, 2364 CrossRef CAS.
-
(a)
Pyrroles, The Synthesis, Reactivity, and Physical Properties of Substituted Pyrroles, Part II; R. A. Jones, ed.; Wiley: NewYork, 1992 Search PubMed;
(b)
D. St. C. Black, Comprehensive Heterocyclic Chemistry II; A. R. Katritzky, C. W. Rees and E. F. V. Scriven, ed.; Elsevier: Oxford, 1996; Vol. 2, p. 39 Search PubMed.
-
(a) C. Rochais, V. Lisowski, P. Dallemagne and S. Rault, Bioorg. Med. Chem., 2006, 14, 8162 CrossRef CAS;
(b) R. Di Santo, R. Costi, M. Artico, G. Miele, A. Lavecchia, E. Novellino, A. Bergamini, R. Cancio and G. Maga, Chem Med Chem., 2006, 1, 1367 CrossRef CAS;
(c) L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS;
(d) B. R. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, Org. Lett., 2006, 8, 701 CrossRef CAS;
(e) G. La Regina, R. Silvestri, M. Artico, A. Lavecchia, E. Novellino, O. Befani, P. Turini and E. Agostinelli, J. Med. Chem., 2007, 50, 922 CrossRef CAS.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1884, 17, 1635 CrossRef.
- C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367 CrossRef.
- A. Hantzsch, Ber. Dtsch. Chem. Ges., 1890, 23, 1474 CrossRef.
-
(a) A. V. Kelin, A. W. Sromek and V. Gevorgyan, J. Am. Chem. Soc., 2001, 123, 2074 CrossRef CAS;
(b) J. T. Kim and V. Gevorgyan, Org. Lett., 2002, 4, 4697 CrossRef CAS;
(c) B. Gabriele, G. Salerno and A. Fazio, J. Org. Chem., 2003, 68, 7853 CrossRef CAS;
(d) D. J. Gorin, N. R. Davis and F. D. Toste, J. Am. Chem. Soc., 2005, 127, 11260 CrossRef CAS;
(e) K. Hiroya, S. Matsumoto, M. Ashikawa, K. Ogiwara and T. Sakamoto, Org. Lett., 2006, 8, 5349 CrossRef CAS;
(f) O. V. Larionov and A. de Meijere, Angew. Chem., 2005, 117, 5809 CrossRef;
(g) O. V. Larionov and A. de Meijere, Angew. Chem., Int. Ed., 2005, 44, 5664 CrossRef CAS;
(h) R. Martin, M. R. Rivero and S. L. Buchwald, Angew. Chem., 2006, 118, 7237 CrossRef;
(i) R. Martin, M. R. Rivero and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 7079 CrossRef CAS.
-
(a) A. D. Omling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168 CrossRef;
(b)
J. Zhu and H. Bienaym'e, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed;
(c) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef CAS;
(d) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef CAS.
- S. Maiti, S. Biswas and U. Jana, J. Org. Chem., 2010, 75, 1674 CrossRef CAS.
-
(a) G. R. Reddy, T. R. Reddy, S. C. Joseph, K. S. Reddy, L. S. Reddy, P. M. Kumar, G. R. krishna, C. M. Reddy, D. Rambabu, R. Kapavarapu, C. Lakshmi, T. Meda, K. K. Priya, K. V. L. Parsa and M. Pal, Chem. Commun., 2011, 47, 7779 RSC;
(b) G. R. Reddy, T. R. Reddy, S. C. Joseph, K. S. Reddy, L. S. Reddy, P. M. Kumar and M. Pal, RSC Adv., 2012, 2, 3387 RSC.
- S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227 CrossRef CAS.
-
(a) F. Epifano, C. Pelucchini, O. Rosati, S. Genovese and M. Curini, Catal. Lett., 2011, 141, 844 CrossRef CAS;
(b) S. Genovese, F. Epifano, M. C. Marcotullio, C. Pelucchini and M. Curini, Tetrahedron Lett., 2011, 52, 3474 CrossRef CAS;
(c) L.-M. Wang, J. Sheng, L. Zhang, J.-W. Han, Z.-Y. Fan, H. Tian and C.-T. Qian, Tetrahedron, 2005, 61, 1539 CrossRef CAS;
(d) A. Kumar and V. K. Rao, Synlett, 2011, 2157 CrossRef CAS;
(e) G.-Y. Lian, F. Lin, J.-D. Yu and D. -W. Zhang, Synth. Commun., 2008, 38, 4321 CrossRef CAS.
-
(a) P. M. Kumar, K. S. Kumar, P. K. Mohakhud, K. Mukkanti, R. Kapavarapu, K. V. L. Parsa and M. Pal, Chem. Commun., 2012, 48, 431 RSC;
(b) K. S. Kumar, P. M. Kumar, M. A. Reddy, M. Ferozuddin, M. Sreenivasulu, A. A. Jafar, G. R. Krishna, C. M. Reddy, D. Rambabu, K. S. Kumar, S. Pal and M. Pal, Chem. Commun., 2011, 47, 10263 RSC;
(c) K. S. Kumar, P. M. Kumar, K. A. Kumar, M. Sreenivasulu, A. A. Jafar, D. Rambabu, G. R. Krishna, C. M. Reddy, R. Kapavarapu, K. Shivakumar, K. K. Priya, K. V. L. Parsa and M. Pal, Chem. Commun., 2011, 47, 5010 RSC;
(d) G. R. Reddy, T. R. Reddy, S. C. Joseph, K. S. Reddy, L. S. Reddy, P. M. Kumar, G. R. Krishna, C. M. Reddy, D. Rambabu, R. Kapavarapu, C. Lakshmi, T. Meda, K. K. Priya, K. V. L. Parsa and M. Pal, Chem. Commun., 2011, 47, 7779 RSC;
(e) T. R. Reddy, G. R. Reddy, L. S. Reddy, S. Jammula, Y. Lingappa, R. Kapavarapu, C. M. T. Lakshmi, K. V. L. Parsa and M. Pal, Eur. J. Med. Chem., 2012, 48, 265 CrossRef;
(f) P. M. Kumar, K. S. Kumar, S. R. Poreddy, P. K. Mohakhud, K. Mukkanti and M. Pal, Tetrahedron Lett., 2011, 52, 1187 CrossRef CAS;
(g) T. R. Reddy, L. S. Reddy, G. R. Reddy, V. S. Nuthalapati, Y. Lingappa, S. Sandra, R. Kapavarapu, P. Misra and M. Pal, Bioorg. Med. Chem. Lett., 2011, 21, 6433 CrossRef.
- Crystallographic data (excluding structure factors) for 4m have been deposited with the Cambridge Crystallographic Data Center as supplementary publication number CCDC 867480†.
- F. Epifano, S. Genovese and M. Curini, Tetrahedron Lett., 2007, 48, 2717 CrossRef CAS.
- S. N. Murthy, B. Madhav, A. V. Kumar, K. R. Rao and Y. V. D. Nageswar, Helv. Chim. Acta, 2009, 92, 2118 CrossRef CAS . While formation of regioisomeric 1,2,3,5-tetrasubstituted pyrroles has been reported in this paper, a comparison of NMR data of compound 4a (isolated from the present MCR) with that of the corresponding regioisomer reported earlier remained inconclusive as both of them appeared to be potentially the same isomer. We thank one of the reviewers for bringing this point to our notice.
-
(a) A. Kodimuthali, S. S. L. Jabaris and M. Pal, J. Med. Chem., 2008, 51, 5471 CrossRef CAS;
(b) S. G. Dastidar, D. Rajagopal and A. Ray, Curr. Opin. Invest. Drugs, 2007, 85, 364 Search PubMed.
- A. Robichaud, P. B. Stamatiou, S.-L. C. Jin, N. Lachance, D. MacDonald, F. Laliberté, S. Liu, Z. Huang, M. Conti and C.-C. Chan, J. Clin. Invest., 2002, 110, 1045 CAS.
- D. Spina, Br. J. Pharmacol., 2008, 155, 308 CrossRef CAS.
- P. Srivani, D. Usharani, E. D. Jemmis and G. N. Sastry, Curr. Pharm. Des., 2008, 14, 3854 CrossRef CAS.
- P. Wang, J. G. Myers, P. Wu, B. Cheewatrakoolpong, R. W. Egan and M. M. Billah, Biochem. Biophys. Res. Commun., 1997, 234, 320 CrossRef CAS.
- J. Demnitz, L. LaVecchia, E. Bacher, T. H. Keller, T. Muller, F. Schurch, H. P. Weber and E. Pombo-Villar, Molecules, 1998, 3, 107 CrossRef CAS.
- Bruker SADABS V2008-1, Bruker AXS.: Madison, WI, USA, 2008.
-
G. M. Sheldrick, SHELX-97, Program for Crystal Structure Determination, University of Göttingen, 1997 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Results of docking studies, copies of NMR spectra for all new compounds. CCDC 867480. For ESI see DOI: 10.1039/c2ra21343e/ |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.