Carbon-supported Ni catalysts with enhanced metal dispersion and catalytic performance for hydrodechlorination of chlorobenzene†
Jia
Wang
,
Guoli
Fan
and
Feng
Li
*
State Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, P. O. BOX 98, Beijing, 100029, P. R. China. E-mail: lifeng@mail.buct.edu.cn; Fax: +86-10-64425385; Tel: +86-10-64451226
First published on 23rd August 2012
Abstract
Novel, highly-dispersed, carbon-supported nickel catalysts with Ni loading from 6.8 to 20.2 wt% were successfully prepared via in situ self-reduction of hybrid Ni–Al layered double hydroxide/carbon (NiAl-LDH/C) nanocomposite precursors, which were assembled by a separate nucleation and aging process accompanied by the carbonization of glucose. The results demonstrated that the carbon component in NiAl-LDH/C precursors serving as the reducing agent could in situ reduce Ni2+ species to Ni0 upon heating under an inert atmosphere, and that such hybrid structures of NiAl-LDH crystals and carbon matrix facilitated the formation of highly-dispersed and uniform Ni nanoparticles with small diameter and large metal surface area. The composition and structure of supported Ni catalysts was profoundly affected by the composition of the NiAl-LDH/C precursors. The as-synthesized supported catalyst with a Ni loading amount of 18.7 wt% displayed excellent catalytic performance in the liquid phase hydrodechlorination of chlorobenzene, giving 99.3% conversion. It was concluded that the dispersion and loading amount of nickel for supported Ni-based catalysts were both critical aspects responsible for the hydrodechlorination activity.
Introduction
It is well known that chlorine-containing effluent discharge has a direct adverse impact on stratospheric ozone, ecosystems, and public health. Nowadays, the development of effective procedures for the hydrodechlorination (HDC) of chloroaromatic compounds is gaining increasing significance as a potential methodology for treating toxic halogenated waste. Compared with conventional thermal incineration, the catalytic HDC of chlorinated organic waste can reuse the valuable raw materials under mild conditions with limited toxic emissions (e.g. polychlorodibenzodioxins and polychlorodibenzofurans) and no CO2 release into the environment.1 In the past decade, many efforts have been focused on the gas phase catalytic HDC of chlorobenzene (CB) and its derivatives,2–15 where the commonly used catalysts are supported noble metal (e.g., Pt, Rh, or Pd)2,3,13–15 or nickel catalysts5–12 due to their good catalytic activity. Meanwhile, the liquid phase HDC of CB on supported nickel-based catalysts has been scarcely reported in the literature.16,17 However, the high cost of noble metal-containing catalysts greatly limits their large-scale practical applications. On the other hand, the most widely used methods for preparing these catalysts are incipient wetness impregnation and direct synthesis. In most cases, these methods usually lead to the formation of large metal particles with poor metal dispersion or relatively low loading amounts,9,12 which originates from the inhomogeneous distribution of active precursors on supports and the weak interactions between them. At present, the design of efficient and recoverable supported Ni-based catalysts has become an important issue in terms of economic and environmental sustainability. As a result, it is imperative to develop green, cheap, and facile methods for the preparation of highly-dispersed, supported Ni nanoparticles with high metal loading amounts.Layered double hydroxides (LDHs), represented by the general formula [M1−x2+Mx3+(OH)2]x+[Ax/n]n−·mH2O, belong to a family of highly ordered two-dimensional layered materials,18 where different M2+ and M3+ metal cations uniformly distribute and orderly prearrange in the brucite-like sheets, and various charge-compensating anions (An−) are present in the interlayer space.19 According to the fact that LDHs can accommodate a large number of tunable M2+ and M3+ ions within the layers or in the interlayer space in the form of metal complexes, well-dispersed supported metal catalysts can be obtained by reducing calcined LDHs with the desired active metal species.20–23 This kind of structural transformation endows LDH materials with an extraordinary capability as catalyst precursors in various metal-catalyzed reactions.24–27 In comparison with other metal-supported catalysts, LDH-derived supported metal catalysts have two advantages: (i) active components with adjustable content can be uniformly integrated into the LDH structure; (ii) metal nanoparticles with tunable particle size can be formed in a controllable manner. Recently, we reported the formation of carbon supported nickel catalysts by reduction of NiAl-LDH/carbon composites (NiAl-LDH/C), which were prepared by the crystallization of NiAl-LDH via a traditional coprecipitation method and the simultaneous carbonization of glucose under hydrothermal conditions. The carbon in the composite serving as a reducing agent could in situ reduce the Ni species to form metallic Ni nanoparticles on a carbon matrix.28
In our previous work, we have adopted a synthetic approach for LDH materials with uniform crystallite size,29,30 which involved a rapid mixing of the two solutions (metal salts and bases) and a nucleation process in the colloid mill for a short period of several minuets. The resultant hydroxide slurry was subsequently aged. Thus, all the particles precipitated out almost at the same time. Thus, nanoparticles with a narrow size range were obtained. Inspired by the above results, in this contribution we established a new synthesis of dispersion-enhanced, supported Ni/C catalysts via in situ self-reduction of hybrid NiAl-LDH/C nanocomposites, which were assembled by a separate nucleation and aging process accompanied by the carbonization of glucose. We have systematically characterized the structure of the composite precursors and the resultant supported catalysts using a combination of chemical analysis, X-ray diffraction (XRD) analysis, in situ high-temperature X-ray diffraction analysis, thermogravimetric–differential thermal analysis (TG-DTA), scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), temperature programmed decomposition and desorption experiments, N2 adsorption–desorption isotherms, and vibrating sample magnetometer measurements, which have endowed us with a further understanding of the relationship between the structures of supported Ni catalysts and their catalytic properties in the liquid phase HDC of CB. Moreover, the as-synthesized supported Ni catalysts combine the advantages of being easy to separate and recover using an external magnetic field, reduced inclination to cause loss or pollution, and excellent stability.
Results and discussion
Characterization of hybrid xNiAl-LDH/C composites
Fig. 1 shows the XRD patterns of the hybrid xNiAl-LDH/C composites. All samples display three characteristic reflections at 2θ angles below 40° attributed to the (003), (006), and (012) planes of hydrotalcite-like materials (JCPDS No. 15-0087) and a characteristic line at a high 2θ of about 62° by the overlapped (110) and (113) planes,31 which are the same as those for the pristine 2.5NiAl-LDH sample (Inset in Fig. 1). Compared with 2.5NiAl-LDH, the broader and weaker characteristic reflections for the xNiAl-LDH/C samples can be attributed to the dilution effect of the resultant carbon component in the composites and the reduced crystalline nature of the LDH phase. Moreover, no reflections related to graphite or other forms of carbon appear, indicative of the presence of carbon in a noncrystalline form. It is believed that under the present hydrothermal treatment, the aromatization and carbonization of glucose lead to the formation of amorphous carbon, and thus the resultant carbon is further assembled with LDH crystallites, leading finally to the formation of a hybrid NiAl-LDH/C composite.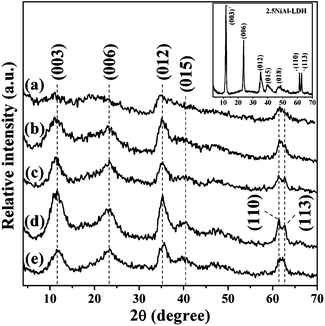 | ||
| Fig. 1 XRD patterns of samples: (a) 1NiAl-LDH/C, (b) 1.5NiAl-LDH/C, (c) 2NiAl-LDH/C, (d) 2.5NiAl-LDH/C and (e) 3NiAl-LDH/C. Inset shows the XRD pattern of the pristine 2.5NiAl-LDH sample. | ||
The SEM image shows that the representative 2.5NiAl-LDH/C composite is constructed of small aggregated particles (Fig. 2a). Furthermore, the TEM observation clearly reveals that the sample presents an interwoven hybrid structure consisting of uniform platelet-like LDH nanoparticles and a noncrystalline carbon component (Fig. 2b), which should be related to the formation of uniform LDH nuclei via novel separate nucleation and aging processes.
 | ||
| Fig. 2 (a) SEM and (b) TEM images of representative 2.5NiAl-LDH/C. | ||
The in situ HT-XRD patterns of the representative 2.5NiAl-LDH/C sample are shown in Fig. 3. A progressive decrease in the relative intensity of characteristic (00l) reflections from 25 to 125 °C is ascribed to the removal of physisorbed water.32 The evident decease in intensity of the (003) peak with increasing temperature is observed within the range of 150–325 °C. Meanwhile, the position of the (003) reflection shifts to a higher 2θ value with the increasing temperature, which is associated with the release of interlayer water, thus giving rise to the collapse of brucite-like layers to form layered double oxide (NiAl-LDO). Above 350 °C, the layered structure of LDH diminishes progressively via both the dehydroxylation of brucite-like layers and the decomposition of interlayer anions. With increasing the temperature to 400 °C, a dominant NiO phase appears.33 On elevating the temperature to 450 °C, the main contribution of reflections at 44.5, 51.8, and 76.4° arises from the (111), (200), and (220) planes of fcc metallic Ni phase (JCPDS No. 04-0850), respectively. The intensities of reflections for metallic Ni phase increase progressively with the increasing temperature, thus demonstrating that upon heating in the absence of H2, the reduction of Ni2+ to Ni0 apparently occurs because carbon in the hybrid composite can also serve as a reducing agent.
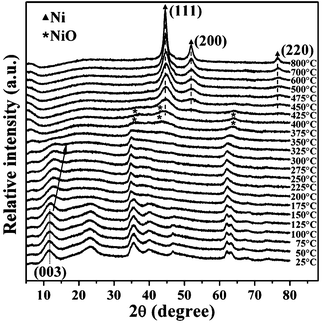 | ||
| Fig. 3 HT-XRD patterns of the 2.5NiAl-LDH/C sample in N2 atmosphere at different temperatures. | ||
Further, the thermal evolution of the representative 2.5NiAl-LDH/C sample was studied by TG-DTA-MS analysis. As shown in Fig. 4, the weight loss occurs essentially in two steps below 800 °C: the first one at lower temprature (ca. 50–265 °C) and the second one within a wide temprature range of ca. 265–800 °C, corresponding to two obvious endothermic events with the endothermic peaks at ca. 189 and 400 °C in the DTA curve. As evidenced by the MS results, the first weight loss can be assigned to the desorption of physisorbed water and the removal of interlayer water, which appear at about 120 and 205 °C, respectively. This result matches well with the above HT-XRD results. Meanwhile, the release of relatively small amounts of physisorbed CO2 on the layers can also be observed. The second weight loss involves the initial dehydroxylation of the lattices as clearly reflected by a H2O peak (∼389 °C), the middle release of CO2 due to decomposition of interlayer anions (∼400 °C), and the final release of CO (∼460 and ∼620 °C) and CO2 (∼465 °C) due to reduction of the NiO phase by the carbon component. Finally, the weight loss above 800 °C is due to the slow release of gas from the micropores of the oxide residue on sintering.34
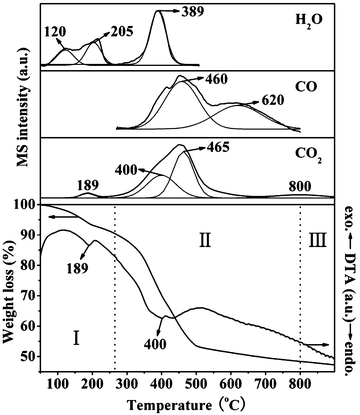 | ||
| Fig. 4 TG-DTA-MS profiles of 2.5NiAl-LDH/C sample. | ||
Characterization of supported Ni/C samples
Fig. 5 shows the XRD patterns of supported Ni/C samples. It is found that the layered structure of the NiAl-LDH phase has disappeared and phase transformation from NiO to Ni is complete at 500 °C. Based on the Scherrer formula, X-ray line-broadening analyses from the (111) reflection of the metallic nickel yield smaller average coherent diffraction domain sizes of Ni nanoparticles in the range of 7.3–8.0 nm for xNi/C samples than for the I–Ni/C sample (about 24.8 nm) (Table 1). Over the course of the synthesis of the composite precursor, NiAl-LDH crystals with nearly uniform size can be obtained in a colloid mill reactor regardless of Ni/Al molar ratios.29 Subsequently, assembly of amorphous carbon with LDH crystallites may take place under the present hydrothermal treatment. As a result, such uniform crystallite size of the metal nanoparticles may result from the high degree of hybrid nanostructure composites, leading to the uniform dispersion of metal species in the samples.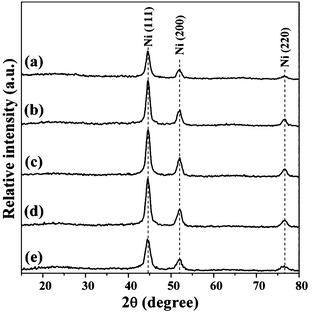 | ||
| Fig. 5 XRD patterns of samples: (a) 1Ni/C, (b) 1.5Ni/C, (c) 2Ni/C, (d) 2.5Ni/C and (e) 3Ni/C. | ||
| Sample | D 111 a (nm) | D b (nm) | S BET c (m2 g−1) | d pore d (nm) | V pore e (cm3 g−1) | Conversionf (%) |
|---|---|---|---|---|---|---|
| a Based on XRD line broadening of (111) plane for Ni metal. b Based on TEM analysis. c BET surface area. d Average pore diameter determined by the BJH method. e Total pore volume. f Reaction time: 200 min. g Average pore diameter and total pore volume determined by the DFT method. | ||||||
| 1Ni/C | 7.9 | 11.2 | 270 | 3.6 | 0.12 | 56.7 |
| 1.5Ni/C | 8.0 | 10.8 | 274 | 4.2 | 0.18 | 65.6 |
| 2Ni/C | 7.5 | 10.4 | 282 | 4.7 | 0.24 | 77.0 |
| 2.5Ni/C | 7.3 | 10.9 | 299 | 8.1 | 0.41 | 99.3 |
| 3Ni/C | 7.5 | 10.7 | 343 | 7.9 | 0.37 | 85.1 |
| I–Ni/C | 24.8 | 31.2 | 154 | 0.55g | 0.07g | 1.2 |
The XPS spectra of the 2.5Ni/C sample are shown in Fig. 6. In the fine spectrum of the Ni 2p region, the peaks at about 852.5 and 859.2 eV are assigned to Ni 2p3/2 and its shake-up satellite, respectively, which is characteristic of metallic Ni species.35 The C 1s spectrum can be deconvoluted into two separate components. The main peak at about 284.5 eV is associated with the sp2-hybridized graphite-like carbon atoms, while the other peak at around 285.6 eV originates from the defects on the graphitic structure. A small peak for carbon π–π* transitions is also present at around 290.8 eV.36 The spectrum of Al 2p shows a peak with binding energy of 74.7 eV, related to the Al3+ species bonded to oxygen,37 suggestive of the formation of amorphous alumina after destruction of NiAl-LDH phase. Correspondingly, amorphous Al2O3 phase attaching to NiO phase over carbon support may further inhibit the aggregation of Ni-containing phases as a dispersant agent during in situ self-reduction, further enhancing the dispersion of Ni nanoparticles.
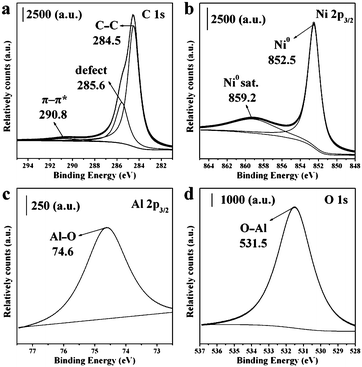 | ||
| Fig. 6 XPS spectra of (a) C1s, (b) Ni 2p3/2, (c) Al 2p3/2 and (d) O1s regions for 2.5Ni/C sample. | ||
TEM analyses were conducted to investigate the morphology and microstructure of Ni nanoparticles of support Ni/C samples with the different loading amounts of Ni (Fig. 7). TEM derived particle size distributions are presented in the insets of Fig. 7a–e, while the extracted surface area weighted mean sizes are summarized in Table 1. TEM micrographs reveal that in the cases of lower Ni loading amounts, Ni nanoparticles are uniformly dispersed on the surface of carbon supports, and exhibit the nature of uniform particle size. In the case of 3Ni/C sample, however, some aggregated nanoparticles are observed, because the higher Ni loading amount in the sample facilitates agglomeration between the nanoparticles. Interestingly, the mean particle sizes with the narrow distribution estimated from TEM micrographs are about 10.4–11.2 nm, in good agreement with that inferred from the XRD results. This demonstrates that Ni nanoparticles have almost uniform particle size in all support Ni/C samples. A typical HRTEM image depicts the lattice fringes with an interplanar spacing of about 1.25 Å for a single nanoparticle (Fig. 7f), corresponding to the (220) plane of fcc metallic nickel phase. Moreover, the relatively thin and faceted nanoparticles of xNi/C are diagnostic of a strong surface interaction between metallic Ni phase and carbon support.38 On the contrary, I–Ni/C sample with the Ni loading amount of 18.7% shows severe aggregation of large globular particles (see ESI†, Fig. S1). The lateral dimension distribution of particles covers a broad range (around 8–42 nm), with an average at 31.2 nm. The above result reveals the nature of poor dispersion of Ni particles and the weak metal–support interaction for the I–Ni/C sample. In tandem with the above experimental observations, a schematic illustration for the formation mechanism of carbon-supported Ni nanoparticles from the NiAl-LDH/C composite precursor is illustrated in Scheme 1.
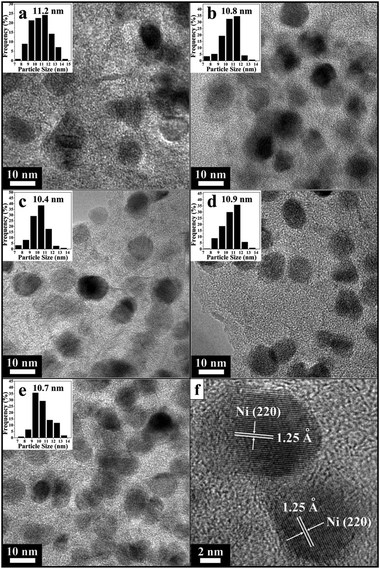 | ||
| Fig. 7 TEM images of samples: (a) 1Ni/C, (b) 1.5Ni/C, (c) 2Ni/C, (d) 2.5Ni/C and (e) 3Ni/C; (f) A typical HRTEM image of 2.5Ni/C with two crystalline Ni nanoparticles on carbon support; insets in (a–e) show histogram of the size distributions of Ni nanoparticles in samples. | ||
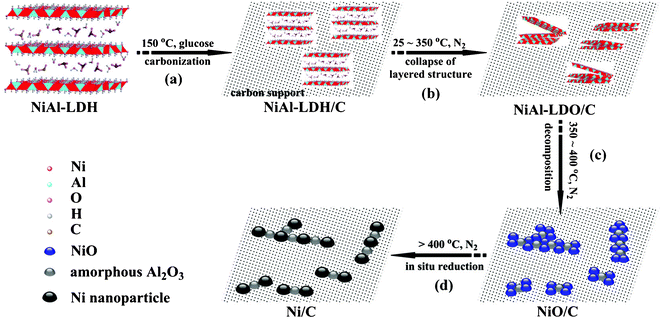 | ||
| Scheme 1 Schematic illustration for the synthesis of carbon-supported Ni nanoparticles (Ni/C). (a) Hybridization of the newly formed NiAl-LDH nanoparticles with carbon generated via carbonization of glucose; (b) formation of NiAl-LDO/C through the collapse of layered structure of NiAl-LDH component; (c) formation of NiO–Al2O3 mixed metal oxides on carbon matrix through structural transformation; (d) formation of Ni/C catalyst via in situ self-reduction by carbon support. | ||
The nitrogen adsorption–desorption isotherms of support Ni/C samples and the corresponding pore-size distribution (PSD) curves are shown in Fig. 8. All the isotherms can be classified as type IV, indicative of the presence of capillary condensation in the mesoporous structure. The 1Ni/C sample shows a H4 hysteresis loop associated with narrow slit-shaped mesopores with uniform size and/or shape.39 With the increasing Ni loading amounts, the hysteresis loop of the samples changes from type H4 to H3. In comparison, 3Ni/C sample presents a H3 hysteresis loop that does not exhibit any limiting adsorption at high P/P0. The H3 hysteresis loop is associated with the aggregates of plate-like particles giving rise to slit-shaped mesopores, indicating that the plate-like morphology of NiAl-LDH precursors is retained in the reduced samples.40 This transformation for the type of hysteresis loop suggests that the increasing number of Ni nanoparticles may result in the formation of a new mesoporous structure, possibly due to a minor accumulation of nanoparticles on the surface of the carbon supports. Therefore, compared with supported Ni/C samples with lower Ni loading amounts, 2.5Ni/C and 3Ni/C samples show a much broader PSD with an additional shoulder peak in the range of 5 to 13 nm (inset of Fig. 8b) and much higher total pore volumes (Table 1), indicative of a well-developed mesoporous structure. In the case of I–Ni/C sample, there is a pronounced adsorption at P/P0 below 10−4 related to the significant micropore structure (see ESI†, Fig. S2). Subsequently, the adsorbed amount rises slowly with the increasing partial pressure. The PSD of I–Ni/C reveals that the sample possesses a large amount of micropores below 1.0 nm and a small amount of mesopores (around 1.5–4.5 nm).
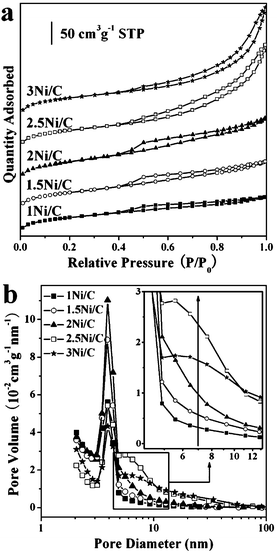 | ||
| Fig. 8 (a) Nitrogen adsorption–desorption isotherms and (b) BJH pore size distribution curves of samples. Inset in (b) shows an enlargement of pore size distribution with the pore diameter from 4–13 nm. | ||
H2-TPD of catalysts is an effective method to illuminate the heterogeneity of catalyst surface and the nature of the interaction between hydrogen species and catalyst surface. It is known that if a non-selective TCD signal in H2-TPD experiments is measured at higher temperatures than those adopted in reduction, there are certainly the signals given by other released gases except for H2. To understand the effect of the released gases produced by the decomposition of supported catalysts on TPD results, TPDE experiments of xNiAl-LDH/C samples were also carried out. Note from Fig. 9A that there is no detective decomposition peak above 500 °C in each case. This indicates that the amount of released gases arising from either decomposition of NiAl-LDH component or carbon support in the hybrid composites is negligible. As a result, TPD measurement in the present system can be used to probe the hydrogen desorption of such supported Ni/C samples. The H2-TPD profiles of samples with different Ni loading amounts are shown in Fig. 9B. Obviously, hydrogen desorption covers a low temperature region of 100–200 °C and a high temperature region above 300 °C. The small desorption peak I below 300 °C is attributed to weak hydrogen desorption on the surface. Since the shapes of the hydrogen desorption peaks at high temperatures are not symmetrical and possess shoulders, each main peak is fitted to two or three peaks (II, III and IV) by deconvoluting the original profiles with the elevated temperature. The intense peak II at relatively low temperature is likely assigned to the desorption of molecular hydrogen bound to the Ni0 species.41 Generally, such hydrogen chemisorbed in molecular form (peak I and II) is believed to be less effective in the hydrogenolytic cleavage of chlorine from the ring.9 The peak III at about 650 °C is attributed to the hydrogen spillover phenomenon.42 The other small desorption peak IV above 700 °C may be attributed to a semi-spillover species associated with the metal/support interface.11 However, hydrogen desorption of the I–Ni/C sample only covers a rather broad temperature region of 100–600 °C (see ESI†, Fig. S3). This suggests a poor and inhomogeneous dispersion of Ni particles for the I–Ni/C sample owing to the presence of larger Ni particles and weaker metal–support interactions.
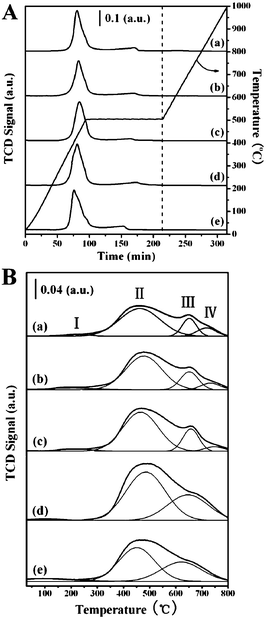 | ||
| Fig. 9 (A) TPDE profiles of samples: (a) 1NiAl-LDH/C, (b) 1.5NiAl-LDH/C, (c) 2NiAl-LDH/C, (d) 2.5NiAl-LDH/C and (e) 3NiAl-LDH/C; (B) H2-TPD profiles of samples: (a) 1Ni/C, (b) 1.5Ni/C, (c) 2Ni/C, (d) 2.5Ni/C and (e) 3Ni/C. | ||
Catalytic performance of supported Ni/C samples
The liquid phase catalytic hydrogenation of CB is typical of a three-phase catalytic reaction, and therefore, several aspects have to be considered in order to achieve a proper and efficient catalyst testing: gas–liquid (G–L), liquid–solid (L–S), and internal diffusion. In the present system, three CNT-supported Ni catalysts do not present both internal and external mass transport limitations in the hydrogenation of CB and thus, the intrinsic activity of the catalysts is actually assessed under our reaction conditions (see ESI†).| Sample | Content (wt.%) | H2 uptakea (mL g−1) | Dispersiona (%) | Initial reaction rate (μmolCl s−1 gNi−1) | TOFb × 102 (s−1) | |
|---|---|---|---|---|---|---|
| Ni | Al | |||||
| a As determined by H2 pulse chemisorption. b Turnover frequency of CB hydrogenation, which was given as the overall rate of CB conversion normalized by the number of active sites over the specified time. | ||||||
| 1Ni/C | 6.8 | 3.1 | 5.3 | 40.5 | 36 | 0.5 |
| 1.5Ni/C | 12.0 | 3.6 | 7.1 | 31.0 | 69 | 1.3 |
| 2Ni/C | 16.9 | 3.8 | 7.9 | 24.5 | 146 | 3.5 |
| 2.5Ni/C | 18.7 | 3.4 | 8.1 | 22.6 | 272 | 7.1 |
| 3Ni/C | 20.2 | 3.0 | 7.8 | 20.2 | 197 | 5.7 |
| I–Ni/C | 18.7 | — | 3.2 | 8.9 | 0.9 | 0.1 |
The catalytic activities of supported Ni/C catalysts in liquid phase HDC of CB are presented in Fig. 10 and Table 2. Note that the catalysts exhibit remarkable differences in the catalytic performance. The conversion of CB over 2.5Ni/C sample increases rapidly within 200 min, and reaches 99.3%, whereas the conversion of CB over 1Ni/C catalyst reaches 22.3% in 200 min. On the one hand, the textural analysis indicates that the 2.5Ni/C and 3Ni/C samples have a well-developed mesoporous structure with relatively larger pore volumes and specific surface areas (Table 1), thus facilitating the mass transfer of reactants in the liquid phase HDC reaction.43 On the other hand, regardless of its lower Ni loading amount, the 2.5Ni/C sample has a higher dispersion of Ni nanoparticles than the 3Ni/C sample (Table 2), as evidenced by TEM and H2 pulse chemisorption results. Therefore, 2.5Ni/C sample possesses more accessible Ni active sites than other supported Ni/C samples.
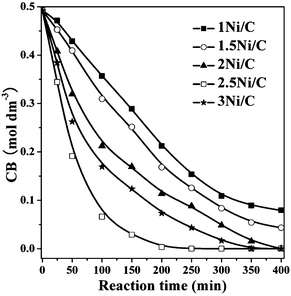 | ||
| Fig. 10 Effect of reaction time on the CB concentration over different catalysts. | ||
Based on the fact that the HDC reaction proceeds via an electrophilic mechanism,44 it is generally presumed that the spillover hydrogen as reactive species, such as H* atoms, radicals,45 H+ ions,46 and H3+ ions,47 should be involved in the hydrogenation. In the present catalyst system, H* and H+ species perhaps coexist on the surface of the as-synthesized Ni/C catalysts.48,49 As shown in Scheme 2, the hydrogen molecules adsorbed on the nickel sites probably undergo homolytic dissociation in the present catalyst system, and the H* atoms produced then spillover onto the metal–support interface to yield a reservoir of surface H+ ions through electron transfer.48 Meanwhile, the Cl groups from CB adsorbed on the catalyst can enhance electron transfer and consequently the amount of strongly held spillover hydrogen.49 Subsequently, HDC takes place via the surface reaction between the spillover hydrogen species and the adsorbed CB. As a result, in the case of the 2.5Ni/C catalyst, the superior catalytic hydrogenation activity should be related to the presence of more accessible Ni active sites originating from appropriate dispersion and loading amount of nickel, as well as strong metal–support interactions, which can give rise to more spillover hydrogen species involved in the hydrogenation process. In contrast, for the I–Ni/C catalyst, poor dispersion of Ni particles and weak metal–support interactions result in the formation of a lesser amount of spillover hydrogen species in the HDC reaction and poor catalytic activity.
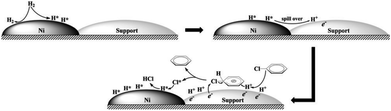 | ||
| Scheme 2 Schematic representation of the hydrodechlorination mechanism of chlorobenzene over carbon-supported Ni catalyst. | ||
Fig. 11 shows the room temperature magnetization curves of the samples. The result shows that the saturation magnetization (Ms) of the supported Ni/C catalysts increases steadily from 10.9 to 22.8 emu g−1 with increasing Ni loading amount. As shown in the inset of Fig. 11, when a magnet with an external magnetic field of 0.15 T approaches the dispersed 2.5Ni/C sample in ethanol at room temperature, particles are attracted to the side wall of the bottle within a relatively short period of 10 s and ethanol becomes clear. When the magnetic field is removed, the 2.5Ni/C sample can be readily re-dispersed in ethanol due to its low remanence and coercivity. This demonstrates the possibility of magnetic separation and recovery for the as-synthesized catalysts using an external magnetic field. Finally, 2.5Ni/C can retain its excellent catalytic activity (reaching the CB yield of 94.1%) after 5 cycles. This implies that the catalyst synthesized by the present method can be almost completely recovered with minimum loss, and is chemically and structurally stable.
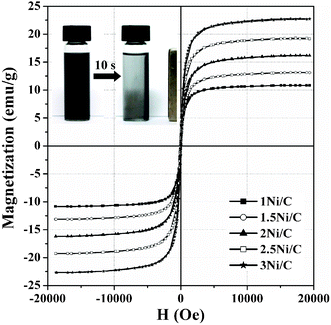 | ||
| Fig. 11 Magnetization curves versus magnetic field for samples at room temperature. Inset shows a photograph representing the magnetic enrichment of the 2.5Ni/C sample in ethanol. | ||
Conclusions
In summary, we report a green route for the synthesis of supported Ni/C catalysts prepared via in situ self-reduction of NiAl-LDH/C nanocomposite precursors, which were assembled through a separate nucleation and aging process accompanied by the carbonization of glucose, using the resulting amorphous carbon as a reducing agent as well as the supporting matrix, in order to stabilize the Ni nanoparticles at a highly-dispersed level. XRD, HT-XRD, and TG-DTA-MS results clearly revealed the reduction of Ni2+ to Ni0 by the carbon component in the composite. TEM, BET, and XPS measurements indicated that the crystallite size of the Ni nanoparticles for the as-synthesized Ni/C catalysts was smaller than that of the catalyst prepared by traditional incipient wetness impregnation. Additionally, the nanocomposite-derived Ni/C catalysts possessed higher nickel dispersion. H2-TPD analyses demonstrated that more spillover hydrogen could be adsorbed on the surface of the 2.5Ni/C sample. Studies of the liquid phase HDC of CB showed that 2.5Ni/C catalyst exhibited excellent catalytic performance with CB conversion up to 99.3%. This is attributed to the presence of more accessible Ni active sites originating from appropriate metal dispersion and loading amount, as well as strong metal–support interactions. The present work provides a simple and feasible synthesis approach for supported Ni-based catalysts with enhanced metal dispersion and catalytic performance. Since the as-synthesized supported Ni catalysts have the advantages of low cost, excellent chemical stability, and facile separation and recovery using a magnetic field, it can be expected that they may have potential applications in the field of heterogeneous catalysis.Experimental
Synthesis of hybrid NiAl-LDH/C composites
Carbonate-containing NiAl-LDH precursors were prepared by a separate nucleation and aging step method. In a typical synthesis, the two solutions were first prepared separately. Solution A: Ni(NO3)3·6H2O and Al(NO3)3·9H2O with the different Ni2+/Al3+ molar ratio x (= 1, 1.5, 2, 2.5, and 3) were dissolved in 30 mL of deionized water to form a clear salt solution ([Al3+] = 0.1 M). Solution B: NaOH and Na2CO3 were dissolved in 30 mL of deionized water to form a mixed base solution ([OH−] = 1.6[Ni2+ + Al3+] and [CO32−] = 2[Al3+]). Then, the solutions A and B were simultaneously added rapidly to a colloid mill with a fixed rotor speed of around 3000 rpm and mixed for 2 min. Subsequently, the suspension was centrifuged, washed, and redispersed in deionized water for 5 cycles to obtain LDH nuclei. Finally, the resulting nuclei were transferred and dispersed into a Teflon-lined autoclave with 30 mL of glucose solution ([C6H12O6] = 2.5[Ni2+ + Al3+]) and aged at 150 °C for 10 h. The resultant suspension was centrifuged and washed with deionized water and ethanol 3 times, respectively. At last, the precipitate was dried at 70 °C to obtain the NiAl-LDH/C hybrid composites (denoted as xNiAl-LDH/C).Synthesis of carbon supported Ni catalysts
The obtained xNiAl-LDH/C composites were placed in a tube-furnace reactor. The furnace was ramped from 30 to 500 °C at a rate of 5 °C min−1 under a nitrogen flow (40 mL min−1), and held at 500 °C for 2 h. Then the furnace was naturally cooled to room temperature. Finally, the black products obtained were denoted as xNi/C. For comparison, a supported Ni catalyst with a Ni loading amount of 18.7 wt.% was prepared by traditional incipient wetness impregnation with nickel nitrate aqueous solution using a carbon support from the carbonization of glucose at 150 °C for 10 h. The slurry was dried overnight at 80 °C, and then the precursor (denoted as Ni(NO3)2/C) was also in situ reduced under a N2 flow (40 mL min−1) at 500 °C for 2 h with a ramping rate of 5 °C min−1. The as-obtained catalyst was denoted as I–Ni/C.Characterizations
XRD patterns of samples were determined on a Shimadzu XRD-6000 diffractometer using Cu-Kα radiation (λ = 0.15418 nm).HTXRD measurements were performed on a PANalytical X′Pert PRO MPD diffractometer (Cu-Kα radiation, X′Celerator detector) connected to an Anton-Paar XRK 900 high-temperature stage. The sample was heated at 5 °C min−1 under N2 atmosphere in steps of 25 °C and stabilized for 5 min before measurements.
Elemental analysis was performed using a Shimadzu ICPS-7500 inductively coupled plasma atomic emission spectroscope (ICP-AES) after the samples were dissolved in dilute nitric acid at 150 °C.
The TG-DTA and mass spectrometry (MS) profiles of the gas evolution were performed on a PerkinElmer Diamond TG/DTA connected to an MS (Pfeiffer ThermoStar). A portion of the volatile products was led to the ion source of the MS through a transfer line heated to 200 °C. The measurements were carried out under an argon atmosphere at a flow rate of 300 mL min−1 with a ramping rate of 5 °C min−1 in a ceramic pan.
X-Ray photoelectron spectroscopy (XPS) was recorded on a VG ESCALAB 2201 XL spectrometer with monochromatic Al Kα X-ray radiation (1486.6 eV photons). The base pressure in the measurement chamber was around 2 × 10−9 Pa. Binding energies were calibrated based on the graphite C 1s peak at 284.5 eV.
Temperature programmed decomposition (TPDE) and H2 temperature programmed desorption (H2-TPD) of the samples were characterized using a Micromeritics ChemiSorb 2720 with a thermal conductivity detector (TCD). For TPDE, the xNiAl-LDH/C sample (100 mg) was reduced in a U-shaped quartz reactor under argon flow (40 mL min−1) at 500 °C (heating rate of 5 °C min−1) for 2 h. Afterward, the temperature linearly increased from 500 to 1000 °C at the same heating rate. For H2-TPD, the xNiAl-LDH/C sample (100 mg) was reduced under the same conditions as for the preparation of the catalyst. Then, Ar was switched to H2/Ar when the sample was cooled to 50 °C. After exposure to 10% v/v H2/Ar for 30 min at 50 °C, the sample was purged by Ar for 2 h at 100 °C in order to eliminate the physical adsorbed H2. Finally, TPD was conducted by ramping from 50 to 800 °C in a stream of argon (40 mL min−1) with a rate of 5 °C min−1. The desorbed hydrogen was monitored continuously by a thermal conductivity detector (TCD). Taking Ag2O as the external standard, the TCD signal was calibrated after removing H2O through a cold trap.
H2 pulse chemisorption was determined using a Micromeritics AutoChem II 2920 instrument. Prior to adsorption measurements, about 200 mg of xNiAl-LDH/C sample was loaded into a U-shaped quartz cell and reduced in a flow of He (50 mL min−1) at 500 °C for 2 h with a heating rate of 5 °C min−1. The temperature was then decreased to 55 °C and subjected to H2 chemisorption using a pulse titration procedure. Hydrogen pulse introduction was repeated until the signal area was constant, indicating surface saturation. The total hydrogen uptake was used to calculate the amount of exposed Ni surface atoms assuming a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H:Ni adsorption ratio.
:1 H:Ni adsorption ratio.
N2 adsorption–desorption isotherms of the samples were obtained on a Micromeritics ASAP 2020 sorptometer apparatus at −196 °C. All samples were outgassed prior to analysis at 200 °C for 12 h under 10−4 Pa vacuum. The total specific surface areas were evaluated with the multipoint Brunauer–Emmett–Teller (BET) method. BET surface areas were reproducible to within ± 5%; the average values were quoted. The mesopore size distribution and average pore diameter were determined by the Barrett–Joyner–Halenda (BJH) method applied to desorption isotherms. The micropore size distribution and mean pore diameter were calculated by the density functional theory (DFT) model applied to adsorption isotherms.
TEM and HRTEM were carried out on a JEOL 2100 operated at an accelerating voltage of 200 kV. The size distribution of particles was determined from more than 500 individual metal particles. The surface area-weighted diameter of particles (D) was calculated from the following equation:
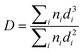 | (1) |
SEM microanalyses of the samples were carried out using a Hitachi S4700 apparatus with an applied voltage of 20 kV.
Magnetism of samples was measured as a function of maximum applied magnetic field to 20 kOe at room temperature on a Lakeshore 7410 vibrating sample magnetometer (VSM).
Catalytic test
The liquid phase catalytic hydrogenation was carried out in a 100 mL stainless steel autoclave. There was no measurable conversion in the absence of the H2 supply. It has been shown previously that the HDC of CB occurs to a significant degree in an excess molar amount of NaOH.50 At the beginning of each experiment, 50 mL of CB solution (0.5 mol dm−3 in ethanol) containing NaOH ([NaOH]/[Cl] = 1.5) was charged with catalyst, where the Cl/Ni ratio was kept at ca. 77 throughout the study according to the experimental results using different molar ratios of NaOH to CB (see ESI†, Fig. S4). Air was then flushed out of the reactor using nitrogen at room temperature. The reactor was then submerged into a water bath at a designated temperature for 2 h under atmospheric pressure. Subsequently, H2 was fed into the reactor at a certain pressure and HDC was initiated by stirring. The total pressure of the autoclave and reaction temperature were kept constant (1.0 MPa and 70 °C) during hydrogenation by supplying hydrogen continuously from gas cylinder through the pressure regulator to compensate for the consumed hydrogen in the autoclave. After reaction, the autoclave was cooled with an ice-water bath for 20 min, and depressurized carefully. Finally, the liquid products were analyzed by an Agilent GC7890A gas chromatograph equipped with flame ionization detector and HP-5 capillary column. The injector temperature was 250 °C, and the detector column temperature was increased from 100 to 150 °C with a ramp rate of 5 °C min−1. Other than CB, no other products were detected by a gas chromatograph mass spectrometer (GCMS-QP2010, Shimadzu). After the reaction, the chlorate ion concentration of produced NaCl was measured by titration with a standard solution of AgNO3 (0.1 M) in the presence of three drops of K2CrO4 (10% w/w) as indicator. The mass balance for chlorine is >98%.Acknowledgements
We gratefully acknowledge financial support from the 973 Program (2011CBA00506) and the National Natural Science Foundation of China.References
- M. A. Keane, G. Pina and G. Tavoularis, Appl. Catal., B, 2004, 48, 275 CrossRef CAS.
- E. J. Creyghton, M. H. W. Burgers, J. C. Jansen and H. van Bekkum, Appl. Catal., A, 1995, 128, 275 CrossRef CAS.
- B. Coq, G. Ferrat and F. Figueras, J. Catal., 1986, 101, 434 CrossRef CAS.
- M. A. Keane and D. Yu. Murzin, Chem. Eng. Sci., 2001, 56, 3185 CrossRef CAS.
- J. T. Feng, Y. J. Lin, D. G. Evans, X. Duan and D. Q. Li, J. Catal., 2009, 266, 351 CrossRef CAS.
- B. F. Hagh and D. T. Allen, Chem. Eng. Sci., 1990, 45, 2695 CrossRef CAS.
- S. Chon and D. T. Allen, AIChE J., 1991, 37, 1730 CrossRef CAS.
- A. R. Suzdorf, S. V. Morozov, N. N. Anshits, S. I. Tsiganova and A. G. Anshits, Catal. Lett., 1994, 29, 49 CrossRef CAS.
- J. Estellé, J. Ruz, Y. Cesteros, R. Fernández, P. Salagre, F. Medina and J. E. Sueiras, J. Chem. Soc., Faraday Trans., 1996, 92, 2811 RSC.
- J. Frimmel and M. Zdražil, J. Catal., 1997, 167, 286 CrossRef CAS.
- E. Shin, A. Spiller, G. Tavoularisa and M. A. Keane, Phys. Chem. Chem. Phys., 1999, 1, 3173 RSC.
- N. Lingaiah, Md. A. Uddin, A. Muto, T. Iwamoto, Y. Sakata and Y. Kusano, J. Mol. Catal. A: Chem., 2000, 161, 157 CrossRef CAS.
- V. I. Simagina, O. V. Netskina, E. S. Tayban, O. V. Komova, E. D. Grayfer, A. V. Ischenko and E. M. Pazhetnov, Appl. Catal., A, 2010, 379, 87 CrossRef CAS.
- T. Hara, T. Kaneta, K. Mori, T. Mitsudome, T. Mizugaki, K. Ebitani and K. Kaneda, Green Chem., 2007, 9, 1246 RSC.
- E. N. Balko, E. Przybylski and F. Vontrentini, Appl. Catal., B, 1993, 2, 1 CrossRef CAS.
- W. Wu, J. Xu and R. Ohnishi, Appl. Catal., B, 2005, 30, 129 CrossRef.
- Z. Wu, M. Zhang, Z. Zhao, W. Li and K. Tao, J. Catal., 2008, 256, 323 CrossRef CAS.
- P. S. Braterman, Z. P. Xu and F. Yarberry, Layered Double Hydroxides, in Handbook of Layered Materials, ed. S. M. Auerbach, K. A. Carrado, and P. K. Dutta, Marcel Dekker, New York, 2004, pp. 373 Search PubMed.
- D. G. Evans and R. C. T. Slade, Struct. Bonding, 2006, 119, 1 CAS.
- F. Li and X. Duan, Struct. Bonding, 2006, 119, 193 CrossRef CAS.
- A. I. Tsyganok, T. Tsunoda, S. Hamakawa, K. Suzuki, K. Takehira and T. Hayakawa, J. Catal., 2003, 213, 191 CrossRef CAS.
- Z. P. Xu, J. Zhang, M. O. Adebajo, H. Zhang and C. Zhou, Appl. Clay Sci., 2011, 53, 139 CrossRef CAS.
- C. Gérardin, D. Kostadinova, B. Coq and D. Tichit, Chem. Mater., 2008, 20, 2086 CrossRef.
- C. Resini, T. Montanari, L. Barattini, G. Ramis, G. Busca, S. Presto, P. Riani, R. Marazza, M. Sisani, F. Marmottini and U. Costantino, Appl. Catal., A, 2009, 355, 83 CrossRef CAS.
- A. Olafsen, Å. Slagtern, I. M. Dahl, U. Olsbye, Y. Schuurman and C. Mirodatos, J. Catal., 2005, 229, 163 CrossRef CAS.
- Z. Yu, D. Chen, M. Rønning, T. Vrålstad, E. Ochoa-Fernández and A. Holmen, Appl. Catal., A, 2008, 358, 136 CrossRef.
- N. Barrabes, D. Cornado, K. Foettinger, A. Dafinov, J. Llorca, F. Medina and G. Rupprechter, J. Catal., 2009, 263, 239 CrossRef CAS.
- Y. Wang, J. Wang, G. Fan and F. Li, Catal. Commun., 2012, 19, 56 CrossRef CAS.
- Y. Zhao, F. Li, R. Zhang, D. G. Evans and X. Duan, Chem. Mater., 2002, 14, 4286 CrossRef CAS.
- J. Wang, Z. Lei, H. Qin, L. Zhang and F. Li, Ind. Eng. Chem. Res., 2011, 50, 7120 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173 CrossRef CAS.
- E. Kanezaki, Inorg. Chem., 1998, 37, 2588 CrossRef CAS.
- C. Vaysse, L. Guerlou-Demourgues and C. Delmas, Inorg. Chem., 2002, 41, 6905 CrossRef CAS PubMed.
- G. S. Thomas, A. V. Radha, P. V. Kamath and S. Kannan, J. Phys. Chem. B, 2006, 110, 12365 CrossRef CAS PubMed.
- A. Śrębowata, W. Juszczyk, Z. Kaszkur, J. W. Sobczak, L. Kępiński and Z. Karpiński, Appl. Catal., A, 2007, 319, 181 CrossRef.
- J. Liu, M. R. Zubiri, B. Vigolo, M. Dossot, Y. Fort, J. J. Ehrhardt and E. McRae, Carbon, 2007, 45, 885 CrossRef CAS.
- N. M. Figueiredo, N. J. M. Carvalho and A. Cavaleiro, Appl. Surf. Sci., 2011, 257, 5793 CrossRef CAS.
- C. A. Bessel, K. Laubernds, N. M. Rodriguez and R. T. K. Baker, J. Phys. Chem. B, 2001, 105, 1115 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- L. Zou, F. Li, X. Xiang, D. G. Evans and X. Duan, Chem. Mater., 2006, 18, 5852 CrossRef CAS.
- A. G. Boudjahem, S. Monteverdi, M. Mercy and M. M. Bettahar, J. Catal., 2004, 221, 325 CrossRef CAS.
- W. C. Conner Jr. and J. L. Falconer, Chem. Rev., 1995, 95, 759 CrossRef.
- L. Chen, K. Yang, H. Liu and X. Wang, Carbon, 2008, 46, 2137 CrossRef CAS.
- G. Yuan and M. A. Keane, Ind. Eng. Chem. Res., 2007, 46, 705 CrossRef CAS.
- D. H. Lenz, W. C. Conner Jr. and J. P. Fraissard, J. Catal., 1989, 117, 281 CrossRef CAS.
- U. Roland, R. Salzer, T. Braunschweig, F. Roessner and H. Winkler, J. Chem. Soc., Faraday Trans., 1995, 91, 1091 RSC.
- D. Bianchi, M. Lacroix, G. M. Pajonk and S. J. Teichner, J. Catal., 1981, 68, 411 CrossRef CAS.
- F. Roessner and U. Roland, J. Mol. Catal. A: Chem., 1996, 112, 401 CrossRef CAS.
- U. Roland, T. Braunschweig and F. Roessner, J. Mol. Catal. A: Chem., 1997, 127, 61 CrossRef CAS.
- Y. Ukisu and T. Miyadera, J. Mol. Catal. A: Chem., 1997, 125, 135 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: additional experimental results. See DOI: 10.1039/c2ra21216a |
| This journal is © The Royal Society of Chemistry 2012 |