Highly selective extraction of the uranyl ion with hydrophobic amidoxime-functionalized ionic liquids via η2 coordination†
Patrick S.
Barber
,
Steven P.
Kelley
and
Robin D.
Rogers
*
Center for Green Manufacturing and Department of Chemistry, The University of Alabama, Tuscaloosa, AL, USA. E-mail: rdrogers@as.ua.edu; Fax: +1 205 348 0823; Tel: +1 205 348 4323
First published on 25th July 2012
Abstract
Hydrophobic, amidoxime-functionalized ionic liquids selectively extract UO22+ from aqueous solution via η2 coordination as demonstrated here with extraction, spectroscopic, and crystallographic studies which prove the amidoxime-uranyl coordination mode and extraction mechanism.
Introduction
The world's oceans contain approximately one thousand times the terrestrial supply of uranium at a consistent 3 parts per billion concentration,1 but to overcome the energetic and economic challenges of the low concentration and variety of interfering ions, one must develop an extractant that is highly selective, cheap, and virtually insoluble under the slightly basic pH and high ionic strength of seawater.2 Since nearly all the uranium is mined in a few countries with terrestrial reserves such as Kazakhstan, Australia, and Canada,3 the United States and other nations are interested in exploiting these seawater reserves.In the 1980s, after studying over 200 adsorbents, it was reported that the amidoxime functional group, RC(NH2)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH), appended to polyacrylonitrile was highly selective towards uranium.4,5 Utilizing this selectivity, the Japanese developed an amidoxime-functionalized nonwoven polyacrylonitrile fabric prepared by radiation-induced graft polymerization which showed the successful uptake of 1 kg of uranium from seawater in a submersion time of 240 days.6
NOH), appended to polyacrylonitrile was highly selective towards uranium.4,5 Utilizing this selectivity, the Japanese developed an amidoxime-functionalized nonwoven polyacrylonitrile fabric prepared by radiation-induced graft polymerization which showed the successful uptake of 1 kg of uranium from seawater in a submersion time of 240 days.6
Though well-studied, the coordination of amidoxime to the uranyl dication is not well understood. There are multiple possible coordination motifs for this interaction, including: monodentate, through the oxime oxygen; bidentate, through the oxime oxygen and the amine nitrogen; or η2, side-on coordination through the N–O oximate bond. With simple acetyl and phenyl derivatives of amidoxime it was shown through single-crystal X-ray diffraction studies that the oxime moiety coordinated to the uranyl dication in a monodentate fashion. However these ligands were zwitterionic due to tautomerization, where the –OH proton transferred to the oxime nitrogen atom7 leading to monodentate coordination through the anionic oxygen.
The η2 coordination mode is particularly interesting as it primarily occurs in softer metals and greatly increases the acidity of the oxime proton.8 Amidoxime resins are selective for uranium in the presence of vastly more concentrated metals, especially alkaline earth cations. Since the adsorption of uranium onto amidoxime resins is hypothesized to occur via the exchange of two protons per uranyl cation,9 the selectivity of amidoxime may stem from the ability of uranyl ions to coordinate in an η2 fashion as opposed to the harder alkaline earth cations. Though hypothesized, at the time of our study no solid-state structural evidence existed for the η2 coordination of amidoximate to a uranyl ion. After we presented this work at a conference,10 we did learn of a concurrent experimental and computational study by Hay and coworkers11 which has now been published and to which we will refer to below. Also, recently published is a cyclic imide dioxime ligand which provides possible evidence towards how a pair of amidoxime ligands within a polymer can coordinate to uranyl tridentate.12
As part of a program to develop new amidoxime-based chitin sorbents prepared via ionic liquid (IL, generally thought of as salts that melt below 100 °C)13 extraction and processing of shrimp shells14,15 to extract uranium from seawater, we sought to incorporate an amidoxime coordination site within a hydrophobic IL and directly explore the fundamental aspects of the coordination and separation of the uranyl cation. Ionic liquids have, of course, been widely investigated for their extraction of actinides and lanthanides,16–18 with varying degrees of success. Many ILs have several advantages over traditional solvents for f-element separations including low vapor pressures, high radiolytic stabilities,19 and generally low flammability. However, the metal ion separations have often been complex, proceeding via a variety of mechanisms including anion-exchange,20 cation-exchange,21 solvation, or even multiple exchange mechanisms.12,22,23 Cation and anion exchange mechanisms are usually followed by the loss or leaching of the IL into the other phase due to the exchange occurring with either the cation or anion of the IL itself.19,20 The design of an IL that participates in either a neutral exchange or simply a proton exchange would be ideal.12–17
Results and discussion
Two amidoxime functionalized ILs were synthesized by a three-step, two-pot reaction in moderately high yields (Scheme 1). The alkylation of 1-methylimidazole with either chloroacetonitrile or 4-chlorobutanenitrile gave the corresponding cyano-derivatized IL intermediates a and b.24 Treating the intermediates with excess hydroxylamine in water yielded ILs as the chloride salts, which then phase-separated with the addition of LiNTf2 as molten bis(trifluoromethane)sulfonamide salts, [AO1mim][NTf2] and [AO2mim][NTf2].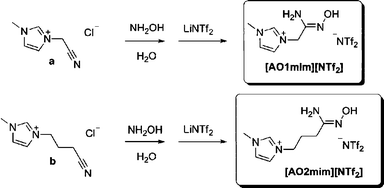 | ||
| Scheme 1 The synthetic scheme for hydrophobic, amidoxime-functionalized ionic liquids. | ||
Thermogravimetric analysis (TGA) of the ILs indicated that they are thermally stable until around 150 °C at which point a small decomposition takes place, 7% and 5% weight loss for [AO1mim][NTf2] and [AO2mim][NTf2], respectively (see Fig. S5†), corresponding to the loss of the oxime moiety from the cation. It is generally reported that amidoximes decompose at around their respective melting points, indicating low thermal stability and supporting the lower decomposition of the oxime moiety within these ILs.25 The second and major decomposition occurs at about 300 °C for both ILs; somewhat lower than other dialkylimidazolium bis(trifluoromethane)sulfonamide salts which decompose between 400–450 °C.26
The mixtures of the ionic liquids with water were found to exhibit upper critical solution temperatures (UCST) of 55 °C and 65 °C for [AO1mim][NTf2] and [AO2mim][NTf2], respectively, above which a completely miscible system is formed. This behavior has been observed before with mixtures of ILs and water, ethanol, or polymers and is useful in separations where the homogenous phases enhance mixing.27–30 Upon cooling, two phases are formed with the lower phase being the IL.
Although two phases can be formed from “hydrophobic” ILs, the mutual solubility of each phase can be quite large (similar to aqueous biphasic systems).31 This partial miscibility can affect the properties, such as viscosity, and potential applications greatly. To quantify the miscibility of the two ILs and water, phase diagrams were constructed by measuring the mole fraction of water in each phase as a function of temperature. After the mixtures were stirred with heating to form a single phase, aliquots of the single phase were cooled to target temperatures. After 24 h of equilibration, the mole fraction of water in each phase was determined by 1H NMR spectroscopy.
Fig. 1 shows the phase diagrams for both ILs which indicate that with increasing temperature increasing amounts of water are incorporated into the IL phase with the longer alkyl chain incorporating less water at the same temperature. For both ILs, the aqueous-rich phases incorporate only very small quantities of the ILs until the miscibility temperature is reached.
![Phase diagrams of [AO1mim][NTf2] (•) and [AO2mim][NTf2] (▼).](/image/article/2012/RA/c2ra21344c/c2ra21344c-f1.gif) | ||
| Fig. 1 Phase diagrams of [AO1mim][NTf2] (•) and [AO2mim][NTf2] (▼). | ||
To test our hypothesis and to confirm the selectivity of the amidoxime ligand for the extraction of uranyl from aqueous solution, we measured the distribution ratios of 233UO22+, 232Th4+, and 152Eu3+ between water and both ILs. Approximately 0.7 g of each IL was mixed with 0.7 mL deionized water and centrifuged to equilibrate the phases. The aqueous solutions were then spiked with the appropriate radiotracer (233UO2Cl2, ca. 0.005 μCi, 232ThCl4, ca. 0.001 μCi, 152EuCl3, ca. 0.01 μCi), mixed, and equilibrated to allow for phase separation. An aliquot of 150 μL of each phase was removed for gamma counting and the distribution ratios were calculated as the counts per minute of the IL phase divided by the counts per minute of the aqueous phase. The extraction of UO22+ was also confirmed by the change in color of each IL to orange after the extraction, along with the loss of the characteristic yellow color of the aqueous solution.
The ILs were both selective for uranyl over either thorium or europium with distribution ratios for [AO1mim][NTf2] of 7.9(4), 1.7(2), and 0.96(2) and for [AO2mim][NTf2] of 24, 2.1(2), and 0.05(1) for UO22+, Th4+, and Eu3+, respectively. These values confirm the selectivity UO22+ > Th4+ > Eu3+ and suggest the quantitative extraction of the uranyl cation is possible with separation factors (SF) of SF(UO2/Th) = 4.6, SF(UO2/Eu) = 8.2, and SF(Th/Eu) = 1.8 for [AO1mim][NTf2] and 11.4, 480, and 42 for [AO2mim][NTf2].
The ionic nature of IL nonaqueous phases gives rise to complicated extraction mechanisms for aqueous metal cations. ILs have only been occasionally shown to extract metal ions in a manner comparable to an organic diluent.32 To explore the extraction mechanism, the distribution ratios were measured as a function of pH and ionic strength. The experiments and calculations were conducted in a manner as described above except the aqueous phase was changed from deionized water to varying concentrations of either HNO3 or NaNO3. Fig. 2 plots the distribution values as a function of [H+] (left) and [NO3−] (right) for [AO1mim][NTf2] and [AO2mim][NTf2].
![Distribution ratios as a function of [H+] (left) and [NO3−] (right) for [AO1mim][NTf2] (•) and [AO2mim][NTf2] (○).](/image/article/2012/RA/c2ra21344c/c2ra21344c-f2.gif) | ||
| Fig. 2 Distribution ratios as a function of [H+] (left) and [NO3−] (right) for [AO1mim][NTf2] (•) and [AO2mim][NTf2] (○). | ||
The distribution values decrease consistently as a function of nitric acid concentration for both ILs, suggesting that the extraction involves deprotonation of the amidoxime. This is based on two considerations. First, deprotonation of the weakly acidic oxime would be suppressed by the addition of the strong acid HNO3. This also explains the difference between the behavior of our system and the more commonly studied (and aprotic) system of n-tributylphosphate (TBP) in dialkylimidazolium33,34 and quaternary ammonium35 [NTf2]− ILs. In these systems the distribution values of uranyl decrease with increasing acid concentration to a point, and then increases again at high concentrations. It is believed that the imidazolium cation is exchanged for cationic uranyl complexes at low acid concentrations, while at higher nitrate concentrations neutral or anionic uranyl complexes are extracted. By contrast, if the IL loses protons in the extraction process it must either extract cationic complexes from or lose anions to the aqueous phase. It therefore stands to reason that the distribution coefficient will not increase at high acid concentrations because the neutral or anionic uranyl nitrate species present will require transfer of the poorly water soluble IL anion into the aqueous phase. This also suggests a convenient stripping methodology to recover the uranium and regenerate the extractant.
The distribution ratios as a function of [NaNO3] were measured to investigate both the role of ionic strength and the role of a competing base in the absence of strong acid. Interestingly, the effects differ for both ILs. The addition of NaNO3 reduces the distribution value of UO22+ into [AO1mim][NTf2], but the distribution values are then constant across various concentrations. However, the distribution values of UO22+ into [AO2mim][NTf2] increase at low concentrations of NaNO3 when compared to deionized water. The distribution values decrease upon increasing the NaNO3 concentration up to 1 M and then increase from 1 to 4 M. These results appear connected with similar examples in the literature suggesting a change in the mechanism at higher nitrate or acid concentrations.36 The higher nitrate content could cause an increase in the water content in the IL or a switch to a cation exchange mechanism with the IL. In both cases this would lead to the increase in the distribution values for the more hydrophobic [AO2mim] IL. The increase in the distribution ratios for [AO2mim][NTf2] may also be due to the fact that the AO2mim+ cation recognizes the shape of the uranyl nitrate complexes (see below).
The pH and 19F-NMR spectra of the aqueous phase were measured after equilibration and after extraction of uranyl from deionized water on a millimolar scale. The aqueous phase did show a 19F peak due to [NTf2]− following equilibration with the IL. However, spectroscopy did not indicate any increase in anion transfer during the extraction. The pH also remained unchanged following the extraction. Since uranyl ions hydrolyze at neutral pH, this suggests that hydroxide anions from species such as [UO2OH]+ accept protons from the AOmim cations.
Single crystal X-ray diffraction was used to obtain information on the coordination of the amidoxime ILs. A single crystal of the neutral complex UO2(NO3)2(AO2mim+−)·H2O was obtained by mixing [AO2mim][NTf2] with uranyl nitrate hexahydrate in methanol and allowing the methanol to evaporate under ambient conditions. In this complex the amidoxime ligand is deprotonated and thus zwitterionic, and the resulting crystalline complex is neutral.
The complex crystallizes in the monoclinic space group P21/n and the asymmetric unit contains an 8-coordinate [UO2(NO3)2(1-(4-amidoximate)butyl)-3-methyl-imidazolium]·complex with distorted hexagonal bipyramidal geometry and an uncoordinated water molecule (Fig. 3). The crystal structure confirms the η2 (O, N) coordination of the amidoximate moiety to the uranyl cation. The presence of uncoordinated lattice water also provides additional evidence that amidoximate coordination is favored over the coordination of water.
![Two views of the hexagonal bipyramidal coordination geometry around uranium in [UO2(NO3)2(1-(4-amidoximate)butyl)-3-methyl-imidazolium]·H2O (50% probability ellipsoids).](/image/article/2012/RA/c2ra21344c/c2ra21344c-f3.gif) | ||
| Fig. 3 Two views of the hexagonal bipyramidal coordination geometry around uranium in [UO2(NO3)2(1-(4-amidoximate)butyl)-3-methyl-imidazolium]·H2O (50% probability ellipsoids). | ||
The six equatorially coordinated atoms and uranium are planar to within 0.03 Å and the UO bond lengths of 1.783(2) and 1.785(2) Å are unremarkable. The U–O(nitrate) bond lengths are in the range 2.455(2)–2.530(2) Å with the nitrate N–O bond lengths being asymmetric. The nitrate N–O(coordinated) bond lengths are in the range 1.263(3)–1.269(5) Å with the nitrate N–O(uncoordinated) bond lengths in the range 1.503(5)–1.513(7) Å. The U–O(amidoximate) and U–N(amidoximate) bond lengths are 2.322(2) and 2.353(3) Å, respectively, with an amidoximate N–O bond length of 1.390(3) Å.
Hay and coworkers have recently reported two amidoximate structures, derivatized by acetyl and phenyl groups.11 Both structures consist of two amidoximate ligands coordinated trans to one another with two methanol molecules completing the coordination sphere. The reported U–O(amidoximate) bond lengths are 2.383(2) and 2.352(2) Å with the U–N(amidoximate) bond lengths 2.398(3) and 2.438(2) Å compared to the much shorter lengths mentioned above. The reported UO bond lengths are slightly longer than those mentioned above at 1.790(2) and 1.796(2) Å.
A Cambridge Structural Database search of all uranyl complexes involving side-on coordination of a deprotonated oximate found twelve crystal structures.37 All structures consist of uranyl ions with equatorial coordination numbers of six. The uranyl complexes are either anionic with three anionic ligands in hexagonal bipyramidal fashion38–41 or neutral with two anionic and two neutral ligands in pseudo-octahedral geometry.42–44 The bond distances of all three reported amidoximate structures are within standard error when compared to the average bond distances of all structures showing side-on oximate coordination (average values of 2.35(6), 2.41(8), and 1.39(5) Å for U–O, U–N, and O–N, respectively).
The neutral complexes with two anionic and two trans ligands have, on average, longer uranyl-oximate bonds than the anionic complexes. Therefore, the structures reported by Hay and coworkers and ourselves have bond lengths and geometries representative of all other crystallographically characterized η2 uranyl oximate complexes. This indicates that the amine group plays a minor role in the coordination.
Interestingly, the coordinated [AO2mim] zwitterion adopts an all-cis conformation of the propyl chain which positions the positively charged imidazolium core directly above one of the coordinated nitrates. This is known to be the most favorable cation–anion interaction site in dialkylimidazolium ILs.45,46 It is unlikely that the [AO1mim] zwitterion, with its single methyl bridge between the amidoximate and the imidazolium ring, would be able to adopt this conformation.
The ability of the [AO2mim] zwitterion to interact in such a manner with the anionic portions of the uranyl complex may explain its much higher distribution ratios in aqueous systems of [AO2mim][NTf2] compared to [AO1mim][NTf2]. This suggests that the bridging moiety (here a propyl group) could be tuned to facilitate favorable interactions between the coordinated anions and the imidazolium ring, increasing selectivity. This might also explain why the addition of nitrate to the initially UO2Cl2-spiked aqueous phase results in a significant increase in the distribution coefficient for [AO2mim][NTf2] but not [AO1mim][NTf2].
The infrared (IR) spectrum of crystalline UO2(NO3)2(AO2mim+−)·H2O was compared to the IR spectrum of a methanol solution containing a mixture of [AO2mim][NTf2] with uranyl nitrate hexahydrate in order to determine if the IL complexes with uranyl in solution. Fig. 4 shows the region of the IR spectra which contains the characteristic asymmetric uranyl UO stretch, which can be used to distinguish the uranyl complexes from each other. This UO stretch in UO2(NO3)2(AO2mim) (886 cm−1) is red-shifted relative to the same stretch in uranyl nitrate hexahydrate (943 cm−1), which is consistent with the replacement of the neutral water molecules by more strongly donating anionic amidoximate ligands. The solution shows a strong IR band at 893 cm−1, indicating that the amidoximate moiety coordinates in solution as well noted by the indicative red shift.
![Infrared spectra comparing [AO2mim][NTf2] (black), [AO2mim][NTf2] with UO2(NO3)2 (red), UO2(NO3)2 hexahydrate (green), and crystals of [UO2(NO3)2(1-(4-amidoximate)butyl)-3-methyl-imidazolium] monohydrate (blue).](/image/article/2012/RA/c2ra21344c/c2ra21344c-f4.gif) | ||
| Fig. 4 Infrared spectra comparing [AO2mim][NTf2] (black), [AO2mim][NTf2] with UO2(NO3)2 (red), UO2(NO3)2 hexahydrate (green), and crystals of [UO2(NO3)2(1-(4-amidoximate)butyl)-3-methyl-imidazolium] monohydrate (blue). | ||
Conclusions
We have synthesized two new amidoxime-functionalized, hydrophobic ionic liquids in a simple three-step process. The phase miscibility with water, extraction behavior, IR data, and solid state characterization support an η2 amidoximate coordination mechanism for the extraction of UO22+ from aqueous solutions. The high distribution ratios for UO22+ provide high selectivity over Th4+ and Eu3+ suggesting these ILs could play a role in the separation of uranium from nuclear waste. In total, this study provides evidence of the extraction mechanism of the amidoxime polymers that have been successfully employed in the extraction of uranium from seawater.Acknowledgements
The authors would like to thank the DOE Office of Nuclear Energy's Nuclear Energy University Programs (Sub-Contract - #120427, Project - #3123).References
-
G. Choppin, J. Rydberg and J.-O. Liljenzin, Radiochemistry and Nuclear Chemistry, 2nd Ed., Butterworth-Heinemann, Great Britain, 1995, p. 107 Search PubMed
.
- R. V. Davies, J. Kennedy, R. W. McIlroy, R. Spence and K. M. Hill, Nature, 1964, 203, 1110 CrossRef
- World Nuclear Organization, Supply of Uranium, http://www.world-nuclear.org/info/inf75.htm (accessed 01/01/11).
- H. J. Schenk, L. Astheimer, E. G. Witte and K. Schwochau, Sep. Sci. Technol., 1982, 17, 1293 CrossRef CAS
- L. Astheimer, H. J. Schenk, E. G. Witte and K. Schwochau, Sep. Sci. Technol., 1983, 18, 307 CrossRef CAS
- M. Tamada, Current Status of Technology for Collection of Uranium from Seawater, Japan Atomic Energy Agency, 2009 Search PubMed
- E. G. Witte, K. S. Schwochau, G. Henkel and B. Krebs, Inorg. Chim. Acta, 1984, 94, 323 CrossRef CAS
- V. Y. Kukushkin, D. Tudela and A. J. L. Pombeiro, Coord. Chem. Rev., 1996, 156, 333 CrossRef CAS
- A. Zhang, T. Asakura and G. Uchiyama, React. Funct. Polym., 2003, 57, 67 CrossRef CAS
- P. S. Barber, S. P. Kelley and R. D. Rogers, Design and Coordination of f-Elements with Amidoxime-Functionalized Ionic Liquids, 17th Symposium on Separation Science and Technology for Energy Applications (Oct. 23–27, 2011), Gatlinburg, TN, Abstract 621 Search PubMed.
- S. Vukovic, L. A. Watson, S. O. Kang, R. Custelcean and B. P. Hay, Inorg. Chem., 2012, 51, 3855 CrossRef CAS
- G. Tian, S. Teat, Z. Zhang and L. Rao, Dalton Trans., 2012 10.1039/c2dt30978e
-
R. D. Rogers and K. R. Seddon, ACS Symposium Series 818, American Chemical Society, Washington D.C., 2002 Search PubMed
- Y. Qin, X. Lu, N. Sun and R. D. Rogers, Green Chem., 2010, 12, 968 RSC
-
Y. Qin, R. D. Rogers and D. T. Daly, Process for forming films, fibers, and beads from chitinous biomass, PCT Int. Appl. (2010), WO 2010141470 A2 12/9/10 Search PubMed
- X. Sun, H. Luo and S. Dai, Chem. Rev., 2012, 112, 2100 CrossRef CAS
- A. E. Visser, R. P. Swatloski, W. M. Reichert, R. Mayton, S. Sheff, A. Wierzbicki, J. H. Davis Jr. and R. D. Rogers, Chem. Commun., 2001, 135 RSC
- I. Billard, A. Ouadi and C. Gaillard, Anal. Bioanal. Chem., 2011, 400, 1555 CrossRef CAS
- D. Allen, G. Baston, A. E. Bradley, T. Gorman, A. Haile, I. Hamblett, J. E. Hatter, M. J. F. Healey, B. Hodgson, R. Lewin, K. V. Lovell, B. Newton, W. R. Pitner, D. W. Rooney, D. Sanders, K. R. Seddon, H. E. Sims and R. C. Thied, Green Chem., 2002, 4, 152 RSC
- M. P. Jensen, J. Neuefeind, J. Beitz, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2003, 125, 15466 CrossRef CAS
- M. L. Dietz, J. A. Dzielawa, I. Laszak, B. A. Young and M. P. Jensen, Green Chem., 2003, 5, 682 RSC
- V. A. Cocalia, J. D. Holbrey, K. E. Gutowski, N. J. Bridges and R. D. Rogers, Tsinghua Sci. Technol., 2006, 11, 188 CrossRef CAS
- V. A. Cocalia, K. E. Gutowski and R. D. Rogers, Coord. Chem. Rev., 2006, 250, 755 CrossRef CAS
- D. M. Drab, M. Smiglak, J. L. Shamshina, S. P. Kelley, S. Schneider, T. W. Hawkins and R. D. Rogers, New J. Chem., 2011, 35, 1701 RSC
- F. Eloy and R. Lenaers, Chem. Rev., 1962, 62, 155 CrossRef CAS
- C. P. Fredlake, J. M. Crosthwaite, D. G. Hert, S. N. V. K. Aki and J. F. Brennecke, J. Chem. Eng. Data, 2004, 49, 954 CrossRef CAS
- L. P. N. Rebelo, V. Najdanovic-Visak, R. Gomes de Azevedo, J. M. S. S. Esperanca, M. Nunes da Ponte, H. J. R. Guedes, Z. P. Visak, H. C. de Sousa, J. Szydlowski, J. N. Canongia Lopes and T. C. Cordeiro, ACS Symp. Ser., 2005, 901, 270 CrossRef CAS
- J. E. L. Dullius, P. A. Z. Suarez, S. Einloft, R. F. de Souza and J. Dupont, Organometallics, 1998, 17, 815 CrossRef CAS
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765 RSC
- H. Rodríguez and R. D. Rogers, Fluid Phase Equilib., 2010, 294, 7 CrossRef
- J. G. Huddleston, S. T. Griffin, J. Zhang, H. D. Willauer and R. D. Rogers, ACS Symp. Ser., 1999, 716, 79 CrossRef CAS
- V. A. Cocalia, M. P. Jensen, J. D. Holbrey, S. K. Spear, D. C. Stepinski and R. D. Rogers, Dalton Trans., 2005, 1966 RSC
- I. Billard, A. Ouadi, E. Jobin, J. Champion, C. Gaillard and S. Georg, Solvent Extr. Ion Exch., 2011, 29, 577 CrossRef CAS
- M. L. Dietz and D. C. Stepinski, Talanta, 2008, 75, 598 CrossRef CAS
- T. Bell and Y. Ikeda, Dalton Trans., 2011, 40, 10125 RSC
- M. L. Dietz and D. C. Stepinski, Talanta, 2008, 75, 598 CrossRef CAS
- F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef
- A. G. Beirakhov, I. M. Orlova, Z. R. Ashurov, G. M. Lobanova, Y. N. Mikhailov and R. N. Shchelokov, Zh. Neorg. Khim., 1990, 35, 3139 CAS
- A. G. Beirakhov, I. M. Orlova, Z. R. Ashurov, G. M. Lobanova, Y. N. Mikhailov and R. N. Shchelokov, Zh. Neorg. Khim., 1991, 36, 654 CAS
- A. G. Beirakhov, I. M. Orlova, Z. R. Ashurov, G. M. Lobanova, Y. N. Mikhailov and R. N. Shchelokov, Zh. Neorg. Khim., 1991, 36, 647 CAS
- A. G. Beirakhov, I. M. Orlova, Y. E. Gorbunova, Y. N. Mikhailov and R. N. Shchelokov, Zh. Neorg. Khim., 1999, 44, 1492 CAS
- A. G. Beirakhov, I. M. Orlova, E. G. Il'in, Y. E. Gorbunova and Y. N. Mikhailov, Zh. Neorg. Khim., 2007, 52, 2011 CAS
- A. G. Beirakhov, I. M. Orlova, E. G. Il'in, Y. E. Gorbunova and Y. N. Mikhailov, Zh. Neorg. Khim., 2007, 52, 39 CAS
- R. Graziani, U. Casellato, P. A. Vigato, S. Tamburini and M. Vidali, J. Chem. Soc., Dalton. Trans., 1983, 697 Search PubMed
- C. Hardacre, J. D. Holbrey, S. E. J. McMath, D. T. Bowron and A. K. Soper, J. Chem. Phys., 2003, 118, 273 CrossRef CAS
- C. Hardacre, J. D. Holbrey, M. Nieuwenhuyzen and T. A. Youngs, Acc. Chem. Res., 2007, 40, 1146 CrossRef CAS
Footnote |
| † Electronic Supplementary Information (ESI) available: details of the synthesis and characterization of the ILs, the metal extraction experiments, and the packing diagrams of the crystal structure. CCDC reference numbers 890413. For ESI and crystallographic data in CIF or other electronic format see DOI:10.1039/c2ra21344c |
| This journal is © The Royal Society of Chemistry 2012 |