A binuclear silver complex with L-buthionine sulfoximine: synthesis, spectroscopic characterization, DFT studies and antibacterial assays†
Fernando Rodrigues Goulart
Bergamini
a,
Marcos Antonio
Ferreira
Jr.
a,
Raphael Enoque Ferraz
de Paiva
a,
Alexandre Ferreira
Gomes
b,
Fábio Cesar
Gozzo
b,
André Luiz Barboza
Formiga
c,
Fabiana Cristina Andrade
Corbi
d,
Italo Odone
Mazali
d,
Danilo Antonini
Alves
e,
Marcelo
Lancellotti
e and
Pedro Paulo
Corbi
*a
aBioinorganic and Medicinal Chemistry Research Laboratory, Institute of Chemistry, University of Campinas—UNICAMP, P.O. Box 6154, 13083-970 Campinas, São Paulo, Brazil. E-mail: ppcorbi@iqm.unicamp.br
bCoordination Chemistry Laboratory, Institute of Chemistry, University of Campinas—UNICAMP, Campinas, São Paulo, Brazil
cDalton Mass Spectrometry Laboratory, Institute of Chemistry, University of Campinas—UNICAMP, Campinas, São Paulo, Brazil
dSolid-State Chemistry Laboratory, Institute of Chemistry, University of Campinas—UNICAMP, Campinas, São Paulo, Brazil
eLaboratory of Biotechnology, Institute of Biology, University of Campinas—UNICAMP, Campinas, São Paulo, Brazil
First published on 3rd September 2012
Abstract
A binuclear silver(I) complex with the amino acid L-buthionine sulfoximine (BSO) of composition Ag2C8H16N2O3S was synthesized and characterized by chemical and spectroscopic measurements, and DFT (density functional theory) studies. Solid-state 13C nuclear magnetic resonance (SSNMR) and infrared vibrational spectroscopy (IR) analyses indicate the coordination of the nitrogen and carboxylate groups of the amino acid moiety to one of the silver atoms, while coordination to the second silver atom occurs through the nitrogen of the sulfoximine group. ESI-QTOF-MS measurements show the maintenance of the binuclear structure in solution. DFT studies confirm the proposed structure as a minimum of the potential energy surface (PES) with calculations of the hessians showing no imaginary frequencies. Raman spectroscopic measurements of the [Ag2(BSO)] complex led to the assignments of the Ag–N bonds. Biological assays of BSO and [Ag2(BSO)] were performed by the well-diffusion method over Staphyloccocus aureus (Gram-positive), Escherichia coli and Pseudomonas aeruginosa (Gram-negative) bacterial strains. The ligand was inactive under the tested concentration (100 μg mL−1). The [Ag2(BSO)] complex was active against the Gram-negative and Gram-positive bacteria tested, with MIC values of 3.125 μg mL−1.
Introduction
Silver compounds have been considered for centuries in the treatment of infectious diseases.1 Although effective, the use of silver salts in high concentrations for the treatment of skin bacterial infections has led to toxic side effects. The fast and uncontrolled release of the metal ion and its further accumulation in the kidneys and liver are probably the most prominent causes of silver intoxications.2 The clinical introduction of sulfonamides in the 1930's, in addition to the serendipitous discovery of penicillin by Sir Alexander Fleming, can be considered a historic event in the treatment of bacterial infections. The discovery of the biological activities of such compounds led to the development of new synthetic and semi-synthetic organic antibacterial drugs.3–5 As a result, the synthesis and biological applications of new silver-based compounds as antibacterial agents were drastically reduced.1Silver nitrate was reconsidered to for clinical use in the 1960's, when Moyer et al. proposed that a 0.5% aqueous silver nitrate solution could be efficiently used on burns against the S. aureus bacterial strain. According to the authors, the use of silver nitrate in this concentration does not interfere in epidermal proliferation.6 This resurgence was closely followed by the discovery of the antibacterial activities of silver(I)-sulfadiazine in 1968.7 Silver(I)-sulfadiazine presents considerable antibacterial activity against both Gram-negative and Gram-positive bacterial strains with reduced adverse effects when compared to silver(I) nitrate.7
In spite of the antibacterial effect of free sulfadiazine, the antibacterial activity of silver(I)-sulfadiazine was reported to be exclusively due to the silver ion, since only it was found inside the bacterial cells. Sulfadiazine was considered responsible for the controlled release of the metal ion.7–11
These results have stimulated the search for new active silver(I) complexes, since silver(I) based antiseptic materials have a low propensity to induce bacterial resistance in comparison to common antibiotics.12 In addition, silver also presents low toxicity when compared to other heavy metals used for the same purpose, such as gold and platinum.12–14
Nowadays, some bacterial strains have been shown to be resistant even to the most effective commercially available antibiotics. For instance, there are meticillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus faecium, extended-spectrum β-lactamase-producing Enterobacteriaceae and multi-resistant Pseudomonas aeruginosa and Acinetobacter baumannii.15 The intrinsic or acquired resistance of some bacterial strains, the latter closely related to the indiscriminate use of antibiotics, led to the development of new antibacterial compounds to overcome multiresistant bacterial strains.16–19 Combining bioactive compounds and metal ions which exhibit bacterial activity is one of the possible approaches against bacterial multiresistance.20,21 The coordination of molecules such as antibacterial agents or aspecific enzyme inhibitor compounds with metal ions such as silver and gold is also desirable due to the possibility to target different bacterial strains in different phases of bacterial growth.22,23
Two silver(I) complexes derived from N-acetyl-L-methionine (L-Hacmet) and N-acetyl-DL-methionine (DL-Hacmet), {[Ag(L-acmet)]}n and {[Ag2(D-acmet)(L-acmet)]}n, were reported to possess a wide spectrum of antimicrobial activities against the Gram-negative E. coli and P. aeruginosa bacterial strains (MIC 15.7 μg mL−1), and the yeasts Candida albicans (MIC 15.7 μg mL−1) and Saccharomyces cerevisiae (MIC 31.3 μg mL−1).24 Cuin et al. recently reported the synthesis of silver(I), gold(I) and gold(III) complexes with 6-mercaptopurine (MP).25 The Ag(I)-MP and Au(I)-MP complexes showed good activity against Mycobacterium tuberculosis, responsible for tuberculosis. In addition, our research group recently reported the synthesis, characterization, DFT studies and preliminary antibacterial assays of Ag(I)-N-acetyl-L-cysteine (Ag-NAC) and Ag(I)-Nimesulide (Ag-NMS) complexes.26,27 In the case of the Ag-NAC complex, coordination of the ligand to Ag(I) was through the sulphur atom, whereas in the Ag-NMS complex coordination of the ligand was through the nitrogen and oxygen atoms of the sulfonamide group. Both compounds were tested as antibacterial agents using the disc diffusion method, being shown to be active against P. aeruginosa, E. coli and S. aureus bacterial strains. Besides, the gold(I) complex with N-acetyl-L-cysteine was also synthesized. Biological studies are in progress in order to compare its antibacterial activities with the respective silver(I) complex.28
L-Buthionine sulfoximine (BSO, C8H18N2O3S, M.W. 222.31 g mol−1) is a specific and potent inhibitor of γ-glutamilcysteine synthetase, which decreases the level of glutathione in tumor cells.29–32 Glutathione is one of the major intracellular antioxidants with multiple biological functions,33 but has the negative aspect of also being related to the development of resistance of several tumors to anti-cancer drugs.34–37 BSO is used as an adjuvant in treatment with metallodrugs such as cis-diamminodichloridoplatinum(II) (cisplatin).38 Besides its biological properties, BSO is a polyfunctional ligand due to the presence of two basic nitrogen atoms, the oxygen in the sulfoximine group and a carboxylic moiety. Here, we describe the synthesis of a new Ag(I) complex with L-buthionine sulfoximine [Ag2(BSO)] in aqueous solution, and its full characterization by elemental, ESI-QTOF mass spectrometric and thermogravimetric (TG) analyses, infrared (IR), Raman and solid-state 13C nuclear magnetic resonance (SSNMR) spectroscopy. Density functional theory (DFT) studies and antibacterial activities of the [Ag2(BSO)] complex against Gram-positive and Gram-negative pathogenic bacterial strains are also described.
Experimental section
Reagents and equipment
L-Buthionine sulfoximine (98%), potassium hydroxide and silver(I) nitrate (AgNO3) (98%) were purchased from Sigma-Aldrich Laboratories. Elemental analyses for carbon, hydrogen and nitrogen were performed using a Perkin Elmer 2400 CHN analyzer. Infrared spectra from 4000–400 cm−1 of BSO and the silver(I) complex [Ag2(BSO)] were measured using an ABB Bomen MB Series FT-IR spectrophotometer; samples were prepared as KBr pellets. The 1H solution-state nuclear magnetic resonance (1H-RMN), 13C solution-state nuclear magnetic resonance (13C-NMR), [1H-13C] heteronuclear single-quantum correlation (HSQC), and [1H-13C] heteronuclear Multiple Bond Correlation (HMBC) spectra of BSO were recorded on a AVANCE III 400 MHz spectrometer, using a 5 mm probe at 303 K. The compound was analyzed in a deuterated water solution. The 13C-{1H} solid-state nuclear magnetic resonance (SSNMR) spectra of BSO and of the [Ag2(BSO)] complex were recorded on a Bruker AVANCE II 400 MHz (9.395T) spectrometer operating at 100 MHz, using the combination of cross-polarization, proton decoupling and magic angle spinning (CP/MAS) at 10 kHz. The 1H radio-frequency field strength was set to give a 90° pulse. Contact time and recycle delay were 4 ms and 1 s, respectively. Samples were analyzed at room temperature and the chemical shifts were referenced to TMS.The 15N NMR chemical shift of BSO was indirectly detected in the solution-state by a heteronuclear [1H–15N] multiple bond coherence (HMBC) experiment. The 1H–15N NMR data were acquired on a Bruker AVANCE III 400 MHz spectrometer, using a 5 mm probe at 303 K. The compound was analyzed in a deuterated water solution. Due to the poor solubility and low percentage of nitrogen in the [Ag2(BSO)] complex composition the 15N NMR data for the complex could not be obtained either in the solution or in the solid-state.
Electrospray ionization quadrupole time-of-flight mass spectrometry (ESI-QTOF-MS) measurements were carried out in a Waters Synapt HDMS instrument (Manchester, UK). A sample of [Ag2(BSO)] was solubilized in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 H2O/MeCN (0.1% formic acid v/v) at a concentration of ca. 4 mg mL−1, then further diluted 100-fold in the same solvent mixture and immediately analyzed. Resulting solutions were directly infused into the instruments ESI source at a flow rate of 15 μL min−1. Typical acquisition conditions were capillary voltage: 3 kV, sampling cone voltage: 20 V, source temperature: 100 °C, desolvation temperature: 200 °C, cone gas flow: 30 L h−1, desolvation gas flow: 900 L h−1, trap and transfer collision energies: 6 and 4 eV, respectively. ESI† mass spectra (full scans) and fragment ion spectra for quadrupole-isolated ions (QTOF-MS/MS) were acquired in reflectron V-mode at a scan rate of 1 Hz. For fragment ion spectrum experiments by collision-induced dissociation (argon as collision gas), the desired ion was isolated in the mass-resolving quadrupole, and the collision energy of the trap cell was increased until sufficient fragmentation was observed. Prior to all analyses, the instrument was externally calibrated with phosphoric acid oligomers (H3PO4 0.05% v/v in 50:50 H2O/MeCN) ranging from m/z 99 to 980. Thermal analysis was performed on a SEIKO EXSTAR 6000 thermoanalyzer to obtain simultaneous TGA/DTA in the following conditions: synthetic air, flow rate of 50 cm3 min−1 and heating rate of 10 °C min−1, from 25 °C to 1100 °C.
:50 H2O/MeCN (0.1% formic acid v/v) at a concentration of ca. 4 mg mL−1, then further diluted 100-fold in the same solvent mixture and immediately analyzed. Resulting solutions were directly infused into the instruments ESI source at a flow rate of 15 μL min−1. Typical acquisition conditions were capillary voltage: 3 kV, sampling cone voltage: 20 V, source temperature: 100 °C, desolvation temperature: 200 °C, cone gas flow: 30 L h−1, desolvation gas flow: 900 L h−1, trap and transfer collision energies: 6 and 4 eV, respectively. ESI† mass spectra (full scans) and fragment ion spectra for quadrupole-isolated ions (QTOF-MS/MS) were acquired in reflectron V-mode at a scan rate of 1 Hz. For fragment ion spectrum experiments by collision-induced dissociation (argon as collision gas), the desired ion was isolated in the mass-resolving quadrupole, and the collision energy of the trap cell was increased until sufficient fragmentation was observed. Prior to all analyses, the instrument was externally calibrated with phosphoric acid oligomers (H3PO4 0.05% v/v in 50:50 H2O/MeCN) ranging from m/z 99 to 980. Thermal analysis was performed on a SEIKO EXSTAR 6000 thermoanalyzer to obtain simultaneous TGA/DTA in the following conditions: synthetic air, flow rate of 50 cm3 min−1 and heating rate of 10 °C min−1, from 25 °C to 1100 °C.
The Raman spectrum was recorded using a Jobin-Yvon T64000 single spectrometer system, equipped with a confocal microscope and a nitrogen-cooled charge-coupled device (CCD) detector. The spectrum was collected using a 633.0 nm (1.5 mW) line of He/Ne laser at room temperature. The sample was analyzed in the solid-state.
Synthesis
The silver(I) complex with BSO was synthesized by the reaction of 2.0 mL of an aqueous solution of silver(I) nitrate (9.0 × 10−4 mol) with 6.0 mL of a freshly prepared aqueous solution of BSO containing 4.5 × 10−4 mol of the ligand. The aqueous AgNO3 solution was added to the BSO solution under magnetic stirring at room temperature followed by the addition of 9.0 × 10−4 mol of KOH. After 40 min of constant stirring, the white solid obtained was vacuum-filtered, washed with cold water and dried in a desiccator over P4O10. Elemental analysis led to the following composition: Ag2C8H16N2O3S. Calcd. for Ag2C8H16N2O3S (%): C, 22.0; H, 3.70; N, 6.42; found (%): C, 22.9; H, 3.34; N, 6.60. The [Ag2(BSO)] complex is insoluble in water, ethanol, methanol, dimethylsulfoxide, acetonitrile, chloroform, acetone and hexane. It is slightly soluble in a mixture of water and acetonitrile (50:50 v/v). The composition of the complex has a molar ratio of 2:1 metal/ligand. No single crystals of the complex were obtained, even after several attempts, in order to perform an X-ray structure determination.
Molecular modeling
Geometry optimizations were carried out using the GAMESS software39 with a convergence criterion of 10−6 a.u. in a conjugate gradient algorithm. The LANL2DZ40 effective core potential was used for silver and the atomic 6-31G(d) basis set41 for all other atoms. Density functional theory (DFT) calculations were performed for BSO and [Ag2(BSO)] using the B3LYP42 gradient-corrected hybrid functional to solve the Kohn–Sham equations with a 10−5 convergence criterion for the density charge. For the BSO zwitterions and [Ag2(BSO)] complex the polarizable continuum model (PCM)43 was used to simulate the effect of water in the geometry optimization. The final geometries were confirmed as a minimum of the potential energy surface (PES) with calculation of the hessians. The harmonic vibrational frequencies and intensities were calculated at the same level of theory with the analytical evaluation of the second derivatives of the energy as a function of the atomic coordinates. The calculated intensities were used to generate the theoretical spectra. Frequencies were scaled by a factor of 0.9614, as recommended by Scott and Radom.44 Simulated vibrational spectra were obtained from the sum of the Lorentzian functions with 20 cm−1 half-bandwidths using the software MOLDEN 4.7.45 Raman intensities for the [Ag2(BSO)] complex were simulated by the numerical differentiation procedure applying an electric field of 2 × 10−3 a.u., as previously reported.46 Frequencies were scaled by a factor of 1.0013, as recommended by Scott and Radom, for low frequencies.44Biological assays
Six pathogenic bacterial strains, E. coli ATCC 25922, P. aeruginosa ATCC 27853, P. aeruginosa 31NM, S. aureus ATCC 25923, S. aureus BEC9393 and S. aureus Rib1 were selected. Stock solutions (10.0 mg mL−1) of BSO and AgNO3 in water, and also a 2.0 mL water suspension containing 20.0 mg of the [Ag2(BSO)] complex were prepared before the experiment. Sufficient inocula of the bacterial strains were added to a 24 multiwell plate until the turbidity equaled 0.5 McFarland (∼1.5 × 10−8 CFU mL−1). Then, 100 μL of the BSO and AgNO3 solutions and 100 μL of the [Ag2(BSO)] suspension were added to the plates. The negative control was obtained by leaving one of the wells of each bacterial strain with no addition of the considered compounds. The minimal inhibitory concentration (MIC) of the [Ag2(BSO)] complex was estimated as recommended by the Clinical and Laboratory Standards Institute (CLSI).47 In this case, the [Ag2(BSO)] complex was submitted to serial dilutions (1:1) in a 96 multiwell plate with 100 μL of each compound/dilution. Samples were transferred to the plates containing the respective bacterial strain (0.5 McFarland) in seven decreasing concentrations.
Results and discussion
Infrared spectroscopic data
The [Ag2(BSO)] infrared (IR) spectrum was analyzed in comparison to that of free BSO. The IR spectra of BSO and [Ag2(BSO)] are provided in Fig. 1.![Infrared spectra of (A) BSO and (B) the [Ag2(BSO)] complex.](/image/article/2012/ra/c2ra21433d/c2ra21433d-f1.gif) | ||
| Fig. 1 Infrared spectra of (A) BSO and (B) the [Ag2(BSO)] complex. | ||
Two bands were related to the asymmetrical and symmetrical NH2 stretching bands of the amino group and were observed in the BSO spectrum at 3230 cm−1 and 3144 cm−1, respectively. The δ(NH2) band is observed in the free ligand spectrum at 1516 cm−1. Also, the IR spectrum of the ligand shows a weak band with a maximum at 2095 cm−1, which is assigned to the combination of the asymmetrical NH3+ bending vibration and the torsional oscillation of the NH3+ group.48 The asymmetrical and symmetrical stretching modes of the carboxylate group are observed at 1618 cm−1 and 1447 cm−1, respectively. The energy difference (Δ) between νas(COO−) and νsym(COO−), which is used as the parameter to evaluate coordination modes of the carboxylate group,49,50 is 171 cm−1. Moreover, another band corresponding to NH3+ is also observed in the IR spectrum of the ligand at 1582 cm−1, which reinforces the existence of the ligand in its zwitterionic form. Characteristic sulfoximine (HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO) bands are observed in the BSO spectrum at 1210 cm−1, and 1012 cm−1, and are assigned as the NS and SO stretching vibrations, respectively.51 The combination of the N–H bending with the SO and NS stretching modes is observed at 1139 cm−1. The other characteristic bands for BSO are the asymmetric and symmetric stretching bands due to the CH2 group, which are observed at 2962 and 2930 cm−1, respectively.
SO) bands are observed in the BSO spectrum at 1210 cm−1, and 1012 cm−1, and are assigned as the NS and SO stretching vibrations, respectively.51 The combination of the N–H bending with the SO and NS stretching modes is observed at 1139 cm−1. The other characteristic bands for BSO are the asymmetric and symmetric stretching bands due to the CH2 group, which are observed at 2962 and 2930 cm−1, respectively.
In the [Ag2(BSO)] spectrum, asymmetrical carboxylate stretching appears at 1582 cm−1, while the symmetrical band is observed at 1390 cm−1. So, in this case, the energy difference (Δ) between the νas(COO–) and νsym(COO–) is 192 cm−1. According to the literature, the larger Δ value for the complex when compared to the ligand suggests a monodentate coordination of the carboxylate group to the metal.50
In the [Ag2(BSO)] spectrum, a broad band in the region 3550–3100 cm−1 is observed. This band can be assigned to the hydrogen bonds between the hydration water molecules and the NH2 group of the [Ag2(BSO)] complex. Hydrogen bonds lead to a poor resolution of the asymmetrical and symmetrical N–H stretching bands. As reported in the literature, the shifting of the N–H stretching mode in the spectrum of the complex, when compared to the spectrum of the ligand can be attributed to the coordination of the amino group of BSO to Ag(I). Moreover, the band referring to the combination of the asymmetrical NH3+ bending vibration and the torsional oscillation of the NH3+ group, observed for BSO, is not present in the [Ag2(BSO)] spectrum, which is also indicative of silver(I) coordination to BSO through the nitrogen of the amino group.
The SN and SO stretching bands in the [Ag2(BSO)] spectrum are observed at 1203 cm−1 and at 1012 cm−1, respectively. The shifting of the SN band suggests the coordination of BSO to the second silver ion through the nitrogen of the sulfoximine. The SN–H bending vibration band is not present in the [Ag2(BSO)] spectrum, which also indicates that the other silver ion is coordinated to the nitrogen of the sulfoximine group. The characteristic bands for the asymmetric and symmetric stretching modes of the CH2 group are observed in the same region as for free BSO.
13C and 15N NMR spectroscopic measurements
The structure of BSO with the hydrogen and carbon atoms numbered is shown in Fig. 2. The solution-state 1H-NMR and 13C-NMR for free BSO are presented in the ESI (#1).† For a more accurate assignment of the hydrogen and carbon atoms, the DEPT135, [1H-13C]-HSQC, [1H-13C]-HMBC and [1H-15N]-HMBC NMR data for the ligand in D2O were also obtained. The [1H-15N]-HMBC, [1H-13C]-HMBC and DEPT 135 spectra are shown in the ESI (#2).† The [1H-13C]-HSQC spectrum is presented in the ESI (#3).†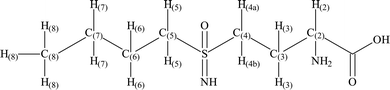 | ||
| Fig. 2 The schematic structure of BSO with the carbon and hydrogen atoms numbered. | ||
As observed in the [1H-13C] HSQC spectrum, the triplet at 0.86 ppm in the 1H-NMR spectrum couples with the carbon at 12.7 ppm in the 13C-NMR spectrum. The high field signals in both the 1H-NMR and 13C-NMR spectra can be respectively attributed to the hydrogen atoms (H-8) and the carbon atom (C-8) of the methyl group. The high field carbons at 20.9 ppm and 23.6 ppm can be assigned, respectively, as C-7 and C-6. The C-7 atom couples with the sextet at 1.39 ppm which can be attributed to the H-7 hydrogen atoms. On the other hand, the C-6 atom couples with the quintet at 1.70 ppm, which is assigned to the H-6 hydrogen atoms. With a chemical shift similar to C-6, the C-3 carbon atom is assigned at 23.6 ppm and couples with the quartet of the H-3 hydrogen atoms at 2.43 ppm. Due to the direct bond of the C-4 to the sulfoximine chiral center, the hydrogen atoms H-4a and H-4b are no longer equivalent and appear in the spectrum as two quartet signals, in the range 3.27–3.36 ppm.
The C-2 and C-5 atoms in the 13C-NMR spectrum appear, respectively, at 53.0 ppm and 53.4 ppm. The C-2 couples with the quartet at 3.80 ppm. The position of C-2 can be explained by the direct bonding of this atom to the primary amine. The C-5 carbon couples with a quartet signal at 3.20 ppm which is assigned to H-5. Finally, the signal that appears at 172.9 ppm is assigned to carbon C-1.
The [1H-13C]-HMBC and [1H-15N]-HMBC spectra confirm the HSQC assignment. The DEPT-135 NMR analysis of BSO confirms C-7, C-6, C-5, C-4 and C-3 as methylenes (CH2).
The NMR spectra of the [Ag2(BSO)] complex were analyzed in comparison to that of BSO. Due to the low solubility of the [Ag2(BSO)] complex in both polar and non-polar solvents, the solid-state nuclear magnetic resonance technique (SSNMR) was applied. The [Ag2(BSO)] complex and the free ligand 13C-{1H} SSNMR spectra are provided in Fig. 3, with the respective carbon assignments.
![The 13C-SSNMR spectra of (A) [Ag2(BSO)] and (B) BSO.](/image/article/2012/ra/c2ra21433d/c2ra21433d-f3.gif) | ||
| Fig. 3 The 13C-SSNMR spectra of (A) [Ag2(BSO)] and (B) BSO. | ||
According to the experimental data, the chemical shift of C-2 in the free ligand spectrum and in the complex spectrum are observed at 53.8 ppm and at 56.6 ppm, respectively, with a Δδ (δ complex ¬ δ ligand) of 2.8 ppm. Moreover, the C-6, C-3 and C-5 signals are observed, respectively, at 23.1 ppm, 26.3 ppm and 56.0 ppm in the free ligand spectrum while for the complex the same carbon atoms appear at 28.9 ppm, 33.7 ppm and 61.6 ppm. The Δδ of the C-6, C-3 and C-5 carbon atoms are ΔδC-6 = 5.8 ppm, ΔδC-3 = 7.4 ppm and ΔδC-5 = 5.6 ppm. As suggested by the IR data, the NMR data reinforces the coordination of BSO to one of the silver atoms through the nitrogen of the sulfoximine. This coordination would lead to changes in the chemical shift of the carbon C-4. In our case, the ΔδC-4 was 0.7 ppm.
In the 13C SSNMR spectra of BSO and the [Ag2(BSO)] complex, C-1 appears as two signals. This phenomenon can be explained due to the existence of polymorphism for both BSO and the [Ag2(BSO)] complex in the solid state.52 The chemical shift for C-1 is observed at 176.6 ppm for the free ligand and at 178.7 ppm for the complex (ΔδC-1 = 2.1 ppm). No substantial difference was observed in the chemical shifts of the C-7 and C-8 carbon atoms in the complex when compared to the free ligand (ΔδC-7 = 1.0 ppm; ΔδC-8 = −0.1 ppm). The carbon assignments for BSO and for the [Ag2(BSO)] complex with their respective Δδ are listed in Table 1. According to the NMR data, we suggest that one of the silver atoms is coordinated to the nitrogen of the amino group and the oxygen of the carboxylate group, while the other silver atom is coordinated to the nitrogen of the sulfoximine group.
| δ (ppm) (BSO) | δ (ppm) (Ag2BSO) | Δδ (ppm) | |
|---|---|---|---|
| C-1 | 176.6 | 178.7 | 2.1 |
| C-2 | 53.8 | 56.6 | 2.8 |
| C-3 | 26.3 | 33.7 | 7.4 |
| C-4 | 52.6 | 53.3 | 0.7 |
| C-5 | 56.0 | 61.6 | 5.6 |
| C-6 | 23.1 | 28.9 | 5.8 |
| C-7 | 22.1 | 23.1 | 1.0 |
| C-8 | 14.5 | 14.4 | −0.1 |
Thermal analysis
Thermogravimetric (TGA) data for the [Ag2(BSO)] complex is presented in the ESI (#4).† Ligand decomposition starts at 170 °C and occurs in two steps, leading to the formation of the residue of the thermal treatment at 550 °C. Calcd. for loss of BSO (%) 48.5; found (%) 53.6. The residue represents the formation of metallic silver. Anal. Calcd. for Ag0 (%) 47.7; found (%) 43.0. Water content (3.44%) is lost until 100 °C. The presence of water is most probably due to absorption from the environment during sample handling before the experiment.Raman spectroscopic measurements
In order to confirm nitrogen coordination of the sulfoximine and of the amino groups to the silver atoms, the Raman spectrum of [Ag2(BSO)] was obtained. The spectrum is provided in the ESI (#5).† As observed, the [Ag2(BSO)] spectrum presents two broad bands with their maxima at 484 cm−1 and 668 cm−1. According to the literature, these bands can be assigned to the Ag–N stretching modes from Ag–N(SO) and Ag–N(H2), respectively.53Mass spectrometry measurements
The Ag2C8H16N2O3S composition was also confirmed by ESI-QTOF-MS measurements. The [Ag2(BSO)] spectrum, presented in Fig. 4, shows the presence of the monoprotonated ion [Ag2BSO−H]+ at m/z 436.91, as well as the presence of the [AgBSO]+ ion at m/z 329.00. The spectrum also shows the presence of the [Ag(BSO)2]+ (m/z 553.11), [Ag2(BSO)2+H]+ (m/z 659.34) and [Ag3(BSO)2−2H]+ (m/z 764.86) ions. The experimental isotopic pattern for [Ag2BSO−H]+ was compared to the expected isotopic pattern considering the proposed composition and was shown to be in good agreement to the latter with an error of −2.0 ppm (calcd. m/z 434.9062, exp. m/z 434.9053).![Mass spectra for the [Ag2(BSO)] complex. (A) The ESI(+)-QTOF mass spectrum from m/z 150 to 800. The term BSO−H refers to the BSO ligand minus one hydrogen (C8H17N2O3S, 221.0960 Da). (B) The isotope pattern comparison for the [Ag2BSO−H]+ ion of m/z 434.90. The mass error was −2.0 ppm for [Ag2BSO−H]+ ([C8H17Ag2N2O3S]+, calcd. m/z 434.9062, exp. m/z 434.9053), considering the monoisotopic ion of the composition.](/image/article/2012/ra/c2ra21433d/c2ra21433d-f4.gif) | ||
| Fig. 4 Mass spectra for the [Ag2(BSO)] complex. (A) The ESI(+)-QTOF mass spectrum from m/z 150 to 800. The term BSO−H refers to the BSO ligand minus one hydrogen (C8H17N2O3S, 221.0960 Da). (B) The isotope pattern comparison for the [Ag2BSO−H]+ ion of m/z 434.90. The mass error was −2.0 ppm for [Ag2BSO−H]+ ([C8H17Ag2N2O3S]+, calcd. m/z 434.9062, exp. m/z 434.9053), considering the monoisotopic ion of the composition. | ||
To further investigate the structure of the observed ions, the [Ag2BSO−H]+ and [AgBSO]+ ions, as well as the [BSO+H]+ ion, were analyzed by ion fragmentation MS/MS spectrometry (Fig. 5). The energy for the fragmentation of the monoprotonated ions was 14 eV for both [BSO+H]+ and [AgBSO]+ and 20 eV for [Ag2BSO−H]+. The [Ag2BSO−H]+ MS/MS spectrum shows a signal at 216.82 m/z, which corresponds to the monoprotonated species minus a C8H16N2O3S fragment. This fragment can be attributed to one BSO ligand with the absence of two hydrogen atoms. The Ag+ ion is also observed in the spectrum at 106.90 m/z.
![The fragment ion mass spectrum (collision-induced dissociation) for the (A) monoprotonated BSO ligand, [BSO+H]+ of m/z 223.11. The collision energy of the trap cell was 14 eV. The term BSO refers to the neutral BSO ligand (C8H18N2O3S, 222.1038 Da). (B) The fragment ion mass spectrum (collision-induced dissociation) for the [AgBSO]+ ion of m/z 329.00. The collision energy of the trap cell was 14 eV. The term BSO refers to the neutral BSO ligand (C8H18N2O3S, 222.1038 Da). (C) The fragment ion mass spectrum (collision-induced dissociation) for the [Ag2BSO−H]+ ion of m/z 436.91. The collision energy of the trap cell was 20 eV. The term [BSO−2H] refers to the BSO ligand minus two hydrogens (C8H16N2O3S, 220.0882 Da).](/image/article/2012/ra/c2ra21433d/c2ra21433d-f5.gif) | ||
| Fig. 5 The fragment ion mass spectrum (collision-induced dissociation) for the (A) monoprotonated BSO ligand, [BSO+H]+ of m/z 223.11. The collision energy of the trap cell was 14 eV. The term BSO refers to the neutral BSO ligand (C8H18N2O3S, 222.1038 Da). (B) The fragment ion mass spectrum (collision-induced dissociation) for the [AgBSO]+ ion of m/z 329.00. The collision energy of the trap cell was 14 eV. The term BSO refers to the neutral BSO ligand (C8H18N2O3S, 222.1038 Da). (C) The fragment ion mass spectrum (collision-induced dissociation) for the [Ag2BSO−H]+ ion of m/z 436.91. The collision energy of the trap cell was 20 eV. The term [BSO−2H] refers to the BSO ligand minus two hydrogens (C8H16N2O3S, 220.0882 Da). | ||
Molecular modeling
The geometries of BSO and [Ag2(BSO)] were obtained by theoretical calculations using density functional theory (DFT). The BSO molecule presents a zwitterionic structure. This structure was optimized and its equilibrium geometry was confirmed by vibrational analysis. The bond distances found for the BSO structure are comparable to those previously reported in the literature for an optimized structure using DFT.54 The theoretical IR spectrum was obtained by calculation of the hessians, showing no imaginary frequencies, and compared to the experimental spectrum, which also confirms the BSO optimized structure in the solid state. For the calculations, the polarizable continuum model (PCM) was employed to account for the effect of water in the description of the zwitterionic forms. For comparative purposes, the [Ag2(BSO)] complex structure was optimized at the same level of theory.As observed by elemental and thermal analysis, the complex presents a 2:1 metal/ligand ratio, which was also observed by the ESI-QTOF-MS analysis. The spectroscopic techniques indicate the coordination of one silver(I) atom through the nitrogen atom of the amino group and a monodentate carboxylate, while the other silver(I) ion appears coordinated to the sulfoximine moiety. So, in the case of the [Ag2(BSO)] complex, the most stable structure is obtained considering the coordination number two for the first silver(I) ion, and coordination number one to the second silver(I) ion, which is bonded to the nitrogen of the sulfoximine group. Silver(I) ions can adopt diverse coordination numbers, passing through coordination numbers one (monodentate) and two to high coordination numbers and geometries, as reported in the literature for various silver compounds.55–58 These data permit us to propose a possible structure for the [Ag2(BSO)] complex, which was also confirmed as a minimum of PES by calculation of the hessians.
The optimized structure for the [Ag2(BSO)] complex is presented in Fig. 6. The calculated Ag–NH2 and Ag–O distances were 2.075 Å and 2.032 Å, respectively, while the H2N–Ag–O angle was 83.0°. The Ag–N and Ag–O distances for the silver atom bonded to the nitrogen atom of the sulfoximine group are 1.992 Å and 2.679 Å. The N–Ag–O angle was 83.1°. Detailed bond distances, angles and dihedrals are reported in the ESI (#6).†
![The [Ag2(BSO)] complex optimized structure obtained by B3LYP/DFT using LANL2DZ(Ag) and 6-31(d,p). The PCM model was used to simulate the water effect on the geometric optimizations.](/image/article/2012/ra/c2ra21433d/c2ra21433d-f6.gif) | ||
| Fig. 6 The [Ag2(BSO)] complex optimized structure obtained by B3LYP/DFT using LANL2DZ(Ag) and 6-31(d,p). The PCM model was used to simulate the water effect on the geometric optimizations. | ||
The simulated and experimental IR spectra of BSO and [Ag2(BSO)] are presented in Fig. 7. The [Ag2(BSO)] IR spectrum is in good agreement with the experimental spectrum. These spectra were used to confirm the experimental assignments. The simulated vibrational spectrum for BSO shows asymmetric amino group stretching νas(H–N–H) at 3461 cm−1 with the symmetric stretching νsym(H–N–H) observed at 3356 cm−1. The characteristic combination of the asymmetrical NH3+ bend vibration and the torsional oscillation of the NH3+ group appear at 2562 cm−1. The difference between the simulated and experimental values for this combination band is larger than usual, but it can be attributed to intermolecular interactions in the solid state.50 The ν(C–O) appears as a combination mode with δ(NH2) at 1345 cm−1. The asymmetric ν(C–O) appears at 1699 cm−1 whereas the asymmetric δ(NH2) appears at 1582 cm−1. A combination mode encompassing the sulfoximine S–N–H bending and the SO and SN stretchings appears at 1128 cm−1. The δ(S–N–H) of the sulfoximine also contributes to the band at 1072 cm−1 whereas the SN stretching is observed at 913 cm−1.
![Simulated infrared spectra of (A) BSO and (C) [Ag2(BSO)]. The experimental spectra of (B) BSO and (D) [Ag2(BSO)] are presented for comparison.](/image/article/2012/ra/c2ra21433d/c2ra21433d-f7.gif) | ||
| Fig. 7 Simulated infrared spectra of (A) BSO and (C) [Ag2(BSO)]. The experimental spectra of (B) BSO and (D) [Ag2(BSO)] are presented for comparison. | ||
In the simulated [Ag2(BSO)] spectrum, the asymmetric and symmetric stretching modes νas(H–N–H) and νsym(H–N–H) appear at 3300 cm−1 and 3186 cm−1, respectively. These data reinforce the loss of the hydrogen atom of the NH3+ group and coordination of the NH2 group to Ag(I).
The bands attributed to the combination mode concerning the sulfoximine SO and SN stretchings appears at 997 cm−1 and 1104 cm−1. When compared to the free BSO spectrum, these bands are shifted −24 cm−1 and −84 cm1, respectively. Moreover, the νas(C–O) band is observed at 1660 cm−1, being shifted −39 cm−1 when compared to the free ligand.
The calculated values for the stretching modes in the Raman spectrum, assigned as N–Ag from silver(I)-amine and silver(I)-sulfoximine are presented in the ESI (#5).† As observed, the Ag–N stretching bands are in good agreement with the experimental data and reinforce the band assignments.
Antibacterial studies
Antibiogram assays were carried out in order to evaluate the antibacterial activities of the Ag-BSO complex. The activities of the complex against the considered bacterial strains were confirmed by MIC values, with concentrations ranging 3.125–100.0 μg mL−1. The results obtained show promising antibacterial activity of the Ag-BSO complex against Gram-negative bacteria, being comparable to the inhibitory effect of the standard antibiotic chloramphenicol, used as positive control. The Ag-BSO complex was less active than the standard antibiotic vancomycin in the MIC assays for Gram-positive S. aureus BEC9393, S. aureus Rib 1 and S. aureus ATCC 25923. The free BSO did not exhibit antibacterial activity under the same experimental conditions. Antibiotic sensitivity profiles of the bacterial strains are listed in Table 2. The observed results show promising potential of application of the Ag-BSO complex as a cream in skin infections in the case of severe burns, due to small number of effective antibiotics against some specific Gram-negative bacterial strains.| Compound | Minimum inhibitory concentration (MIC/μg mL−1) | |||||
|---|---|---|---|---|---|---|
| Gram-negative | Gram-Positive | |||||
| P. aeruginosa ATCC 27853 | P. aeruginosa 31NM | E. coli ATCC 25922 | S. aureus BEC 9393 | S. aureus Rib 1 | S. aureus ATCC 25923 | |
| Ag-BSO | 3.125 | 3.125 | 3.125 | 100 | 100 | 100 |
| Chloramphenicol | 50.0 | 12.5 | 3.125 | — | — | — |
| Vancomycin | — | — | — | <10.0 | <10.0 | <10.0 |
Conclusions
A silver(I) complex with BSO was synthesized and structurally characterized. Elemental, themogravimetric and ESI-QTOF-MS analyses show a 2:1 metal/ligand composition, with the molecular formula [Ag2(C8H16N2O3S)]. The 13C CP/MAS SSNMR, IR and Raman spectroscopic data suggest the coordination of BSO to one silver atom through the amino and carboxylate groups of the amino acid moiety, and also the coordination of the ligand to another silver atom through the nitrogen of the sulfoximine group. DFT studies support the proposed geometry. Biological studies revealed that the complex is effective against all the tested bacteria, being more effective against the tested Gram-negative bacterial strains. Further studies are intended in order to evaluate the possible mechanism of action of the [Ag2(BSO)] complex.
Acknowledgements
This study was supported by grants from the Brazilian Agencies FAPESP (São Paulo State Research Foundation, Brazil—proc. 2006/55367-2, 2008/57805-2, 2012/08230-2 and 2009/54066—Laboratory of Advanced Optical Spectroscopy, LMEOA/IQ—UNICAMP), CAPES and CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Brazil—proc. 573672/2008-3; 472468/2010-3 and 472067/2010-9). The authors are also grateful to MSc. Helen Graci C. de Meneses for her valuable contribution in the DFT studies and to Professor Carol H. Collins for the English revision of the manuscript.References
- H. J. Klasen, Burns, 2000, 26, 117 CrossRef CAS.
- K. H. O. Pelkonen, Heinonen-Tanski Helvi and O. O. P. Hänninen, Toxicology, 2003, 186, 151 CrossRef CAS.
- J. Drews, Science, 2000, 287, 1960 CrossRef CAS.
- A. Fleming, Brit. J. Exp. Pathol., 1929, 10, 226 CAS.
- R. P. Elander, Appl. Microbiol. Biothechnol., 2003, 61, 385 CAS.
- C. A. Moyer, L. Brentano, D. L. Gravens, H. W. Margraf and W. W. Monafo, Arch. Surg., 1965, 90, 812 CrossRef CAS.
- C. L. Fox and S. M. Modak, Antimicrob. Agents Chemother., 1974, 5, 582 CrossRef CAS.
- R. S. Ward and J. R. Saffle, Phys. Ther., 1995, 75, 526 CAS.
- F. C. Tenover, Am. J. Med., 1991, 91, S76 CrossRef.
- C. L. Fox Jr, Arch. Surg., 1968, 96, 184 CrossRef CAS.
- C. L. Fox Jr, Int. Surg., 1975, 60, 275 CAS.
- S. A. Jones, P. G. Bowler, M. Walker and D. Parsons, Wound Repair Regener., 2004, 12, 288 CrossRef.
- Md. S. A. S. Shah, M. Tag, T. Kalagara, S. Singh and S. V. Manorama, Chem. Mater., 2008, 20, 2455 CrossRef CAS.
- Y. Li, X. Dong, Y. Gou, Z. Jiang and H.-L. Zhu, J. Coord. Chem., 2011, 64, 1663 CrossRef CAS.
- G. D. Wright and A. D. Sutherland, Trends Mol. Med., 2007, 13, 260 CrossRef CAS.
- R. Mehrotra, S. N. Shukla, P. Gaur and A. Dubey, Eur. J. Med. Chem., 2012, 50, 149 CrossRef CAS.
- P. R. Murray, K. S. Rosenthal and M. A. Pfaller, Microbiologia Médica, Sixteth ed., Elsevier, Rio de Janeiro, 2009 Search PubMed.
- E. Chartone-Souza, T. L. Loyola, M. Bucciarelli-Rodriguez, M. Ã. B. C. Menezes, N. A. Rey and E. C. Pereira-Maia, J. Inorg. Biochem., 2005, 99, 1001 CrossRef CAS.
- M. Ibrahim, F. Wang, M.-M. Lou, G.-L. Xie, B. Li, Z. Bo, G.-Q. Zhang, H. Liu and A. Wareth, J. Biosci. Bioeng., 2011, 112, 570 CrossRef CAS.
- B. Biersack, R. Diestel, C. Jagusch, F. Sasse and R. Schobert, J. Inorg. Biochem., 2009, 103, 72 CrossRef CAS.
- N. Nikolis, C. Methenitis and G. Pneumatikakis, J. Inorg. Biochem., 2003, 95, 177 CrossRef CAS.
- R. G. de Lima, Transition Met. Chem., 2003, 28, 272 CrossRef.
- S. Pal, E. J. Yoon, S. H. Park, E. C. Choi and J. M. Song, J. Antimicrob. Chemother., 2010, 65, 2134 CrossRef CAS.
- N. C. Kasuga, R. Yoshikawa, Y. Sakai and K. Nomiya, Inorg. Chem., 2012, 51, 1640 CrossRef CAS.
- A. Cuin, A. C. Massabni, G. A. Pereira, C. Q. F. Leite, F. R. Pavan, R. Sesti-Costa, T. A. Heinrich and C. M. Costa-Neto, Biomed. Pharmacother., 2011, 65, 334 CrossRef CAS.
- C. Abbehausen, T. A. Heinrich, E. P. Abrão, C. M. Costa-Neto, W. R. Lustri, A. L. B. Formiga and P. P. Corbi, Polyhedron, 2011, 30, 579 CrossRef CAS.
- R. E. F. de Paiva, C. Abbehausen, A. F. Gomes, F. C. Gozzo, W. R. Lustri, A. L. B. Formiga and P. P. Corbi, Polyhedron, 2012, 36, 112 CrossRef CAS.
- P. P. Corbi, F. A. Quintão, D. K. D. Ferraresi, W. R. Lustri, A. C. Amaral and A. C. Massabni, J. Coord. Chem., 2010, 63, 1390 CrossRef CAS.
- W. B. Rowe and A. Meister, Proc. Natl. Acad. Sci. U. S. A., 1970, 66, 500 CrossRef CAS.
- O. W. Griffith, M. E. Anderson and A. Meister, J. Biol. Chem., 1979, 254, 1205 CAS.
- O. W. Griffith and A. Meister, J. Biol. Chem., 1979, 254, 7558 CAS.
- M. E. Anderson, Chem.-Biol. Interact., 1998, 111–112, 1 CrossRef CAS.
- H.-M. Shen, C.-F. Yang, J. Liu and C.-N. Ong, Free Radical Biol. Med., 2000, 28, 1115 CrossRef CAS.
- S. Ouwerkerk-Mahadevan and G. J. Mulder, Chem.-Biol. Interact., 1997, 105, 17 CrossRef.
- Y. Jia, W. Zhang, H. Liu, L. Peng, Z. Yang and J. Lou, Cancer Chemother. Pharmacol., 2012, 69, 377 CrossRef CAS.
- B. Stordal and M. Davey, IUBMB Life, 2007, 59, 696 CrossRef CAS.
- Y. Lu and A. Cederbaum, Free Radical Biol. Med., 2007, 43, 1061 CrossRef CAS.
- L. Pendyala, R. Perez, A. Weinstein, J. Zdanowics and P. J. Creaven, Cancer Chemother. Pharmacol., 1997, 40, 38 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. K. N. Matsunaga, K. Nguyen, S. Su, T.L. Windus, M. Dupuis and J.A. Montgomery Jr., J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- (a) R. Ditchfie, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS; (c) P. C. Harihara and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef; (d) W. J. Hehre, R. Ditchfie and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117 CrossRef CAS.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502 CrossRef CAS.
- G. Schaftenaar and J. H. Noordik, J. Comput. -Aided Mol. Des., 2000, 14 Search PubMed.
- J. A. Bonacin, A. L. B. Formiga, V. H. S. de Melo and H. E. Toma, Vib. Spectrosc., 2007, 44, 133 CrossRef CAS.
- Clinical and Laboratory Standards Institute (CLSI), Performance Standards for Antimicrobial Susceptibility Testing, Seventeenth Informational Supplement, Wayne, PA, USA, 2007.
- R. M. Silverstein, F. X. Webster, D. J. Kiemle, Spectrometric Identification of Organic Compounds, 7th ed., 2005, John Wiley & Sons, New York, pp. 102–103 Search PubMed.
- P. P. Corbi, M. Cavicchioli, P. Mel'nikov, A. C. Massabni and L. A. A. de Oliveira, Russ. J. Coord. Chem., 2000, 26, 28 CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B, John Wiley & Sons, New York, 5th edn, 1997, pp. 59–62 Search PubMed.
- C. R. Johnson, R. A. Kirchhoff and H. G. Corkins, J. Org. Chem., 1974, 39, 2458 CrossRef CAS.
- M. B. M. Spera, F. A. Quintão, D. K. D. Ferraresi, W. R. Lustri, A. Magalhães, A. L. B. Formiga and P. P. Corbi, Spectrochim. Acta, Part A, 2011, 78, 313 CrossRef.
- B. Morzyk-Ociepa and D. Michalska, Spectrochim. Acta, Part A, 1999, 55, 2671 CrossRef.
- P. S. Kumar and P. V. Bharatam, Tetrahedron, 2005, 61, 5633 CrossRef.
- F. Caruso, M. Camalli, H. Rimml and L. M. Venanzi, Inorg. Chem., 1995, 34, 673 CrossRef CAS.
- R. Noguchi, A. Sugie, A. Hara and K. Nomiya, Inorg. Chem. Commun., 2006, 9, 107 CrossRef CAS.
- N. C. Kasuga, M. Sato, A. Amano, A. Hara, S. Tsuruta, A. Sugie and K. Nomiya, Inorg. Chim. Acta, 2008, 361, 1267 CrossRef CAS.
- M. A. Carvalho, R. E. F. de Paiva, F. R. G. Bergamini, A. F. Gomes, F. C. Gozzo, W. R. Lustri, A. L. B. Formiga, S. M. Shishido, C. V. Ferreira and P. P. Corbi, J. Mol. Struct., 2013, 1031, 125 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2ra21433d |
| This journal is © The Royal Society of Chemistry 2012 |