Formation of well-defined spherical particles during suspension polymerization of biodegradable poly(glycolide-co-p-dioxanone) in supercritical carbon dioxide
Tianqiang
Wang
a,
Jianyuan
Hao
*a,
Yu
Liu
a and
Zhongwei
Gu
*b
aState Key Lab of Electronic Films and Integrated Devices, School of Microelectronics and Solid State Electronics, University of Electronic Science and Technology of China, Chengdu 610054, Sichuan, P R China. E-mail: jyhao@uestc.edu.cn; Fax: 0086-(0)28-83201060; Tel: 0086-(0)28-83201202
bNational Engineering Research Centre for Biomaterials, Sichuan University, Chengdu 610064, Sichuan, P R China. E-mail: zwgu@scu.edu.cn
First published on 3rd September 2012
Abstract
Biodegradable particles of poly(glycolide-co-p-dioxanone) (PGDO) were directly synthesized by suspension copolymerization of glycolide (GA) and p-dioxanone (PDO) in supercritical carbon dioxide (scCO2) using stannous octoate as the ring-opening catalyst and a fluorocarbon polymer surfactant as a stabilizer. Fine powdered products were achieved when more than 60% (w/w) GA was fed. Interestingly, some spherical particles formed when the feed content of GA was intermediate (70–90%), while other compositions resulted in irregular particles. The particle size and degree of aggregation was also affected significantly by copolymer composition. The mechanism for three cases of particle formation were discussed, and the crystallization behavior within the polymerizing droplets played a critical role in determining the final morphology of particles. Well-defined particles with spherical morphology could be achieved if a delicate balance between the two opposing effects of crystallization and plasticization was reached inside the droplets. The possibility that spherical particles for biodegradable polyesters could be fabricated directly by suspension polymerization in scCO2 implies the potential use of this clean technique in biomedical fields.
1. Introduction
Biodegradable polymers are advantageous for use as carriers in biomedical fields due to their known metabolic pathway in the human body and good biocompatibility and biosafety when compared with non-degradable polymers.1,2 They can be processed into different formulations such as films, particles, rods and scaffolds by various techniques. Particle formulations, especially with spherical morphology and well-defined particle size, are of particular interest for use in injected biomedicines.3,4 Good control of spherical morphology is a significant processing requirement for particles because the resulting morphology is related to the stability of the injected suspension solution, well-defined release behavior of guest matter and reproducibility of the formulation.The traditional methods used to process biodegradable polymers into particles involve the use of potentially toxic organic solvents. In recent years, environment-friendly scCO2 has attracted attention as a replacement for traditional organic solvents to prepare biodegradable particles.5,6 scCO2 has many advantages: (1) CO2 is inexpensive, inert, non-toxic, non-flammable and has easily accessible critical parameters (Pc = 73.8 bar/Tc = 31.1 °C); (2) there is a lack of toxic solvent residues in the product and product recovery occurs by simply venting the reaction vessel.7,8 Techniques that have been extensively explored to produce polymeric particles in scCO2 include the rapid expansion of supercritical solution (RESS),9,10 particles from gas-saturated solution (PGSS),11,12 the gas antisolvent process (GAS),13 and the supercritical antisolvent process (SAS)14 and its various modifications, depending on whether the supercritical fluid or dense gas used as a solvent, a plasticizer or an antisolvent.
The combination of polymer synthesis and particle formation in a single step in scCO2 is attractive to generate biodegradable particles to easily produce particles on a large scale. This merged process was realized by suspension or precipitation polymerization of lactide or lactone monomers in scCO2.15–20 So far, biodegradable polymeric particles, such as poly(ε-caprolactone) (PCL), poly(L-lactide) (PLLA), poly(glycolide) (PGA) were produced by polymerization in scCO2in situ. Many studies have shown that the addition of a suitable stabilizer to polymerizations conducted in scCO2 was crucial to acquire powdered products. Stabilizers used in scCO2 typically consisted of a CO2-phobic portion, usually a hydrocarbon polymer that anchored inside or onto the surface of the growing polymer particles, and a CO2-philic segment, such as a fluorinated section, that extended into the CO2 continuous phase preventing particle aggregation by steric stabilization.21
In the case of suspension polymerization, the monomers are dispersed in scCO2 as single droplets, which is different from the dissolution state for precipitation polymerization. Thus, suspension polymerization is a promising technique for the in situ loading of guest matter if it is added into the monomers beforehand. However despite several attempts to successfully achieve powdered products via suspension polymerization, the problem of control over the spherical morphology for the produced particles remained.
In this paper, we demonstrated for the first time that well-defined spherical particles of biodegradable poly(glycolide-co-p-dioxanone) (PGDO) copolymer could be acquired via suspension polymerization of glycolide (GA) and p-dioxanone (PDO) in scCO2. The underlying mechanism for the control of spherical morphology was revealed to be related to a delicate balance between the effects of crystallization and plasticization of the polymerizing droplets. This finding sheds light on the possibility of the technique being used in biomedical fields.
2. Experimental
2.1. Materials
Glycolide (GA) was prepared in our lab and recrystallized from ethyl acetate before use. p-Dioxanone (PDO) was synthesized by well established procedure from ethylene glycol and chloroacetic acid and purified by repeated recrystallization from ethyl acetate to achieve a purity of over 99.5%.22 ε-Caprolactone (CL, Aldrich, 99%) was dried over CaH2 at room temperature for 48 h and distilled under reduced pressure. Telechelic dihydroxyl terminated perfluoropolyether (PFPE) was supplied by Solvay (registered name Fluorolink D10H). The ROP catalyst, tin(II) ethyl hexanoate (Sn(Oct)2; Aldrich) was degassed before use. Research grade CO2, 99.999% purity, was supplied by Guangzhou Puyuan Gas Limited Company. Other reagents were used as received.2.2. Synthesis of the scCO2 soluble PCL-PFPE-PCL stabilizer
The stabilizer synthesis was adapted from that of Pilati et al.23 beginning with ROP of CL from the terminal hydroxyl groups of the “CO2-philic” PFPE macromonomer, in accordance with the activated chain end mechanism proposed by Penczek.24 PFPE (7 g) was reacted in bulk with ε-CL (6.2 mL, 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 PFPE: CL molar ratio) at 150 °C for 18 h using a Sn(Oct)2 (0.3 mL) catalyst. The hydroxyl terminal functionalities were found to initiate the ROP reaction and weaken its solubility in scCO2. Thus, these functionalities were end-capped with acetic anhydride before use to prevent this undesirable side reaction and improve scCO2 solubility. The chemical structure of the stabilizer is shown in Scheme 1.
:1 PFPE: CL molar ratio) at 150 °C for 18 h using a Sn(Oct)2 (0.3 mL) catalyst. The hydroxyl terminal functionalities were found to initiate the ROP reaction and weaken its solubility in scCO2. Thus, these functionalities were end-capped with acetic anhydride before use to prevent this undesirable side reaction and improve scCO2 solubility. The chemical structure of the stabilizer is shown in Scheme 1.
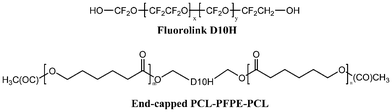 | ||
| Scheme 1 The molecular formation of end-capped PCL-PFPE-PCL. | ||
2.3. Polymerization procedure in scCO2
Polymerization (Scheme 2) was conducted in a 100 ml stainless steel autoclave with a pressure relief valve. Heating was provided via an oil bath, and pressure and temperature were monitored on a digital display control rack. The contents of the reactor were stirred with a Teflon stir bar controlled by a magnetic stir plate. 5 g of monomer, 0.6 ml of catalyst (0.32 g ml−1) and 0.6 g of stabilizer were placed into the reactor prior to pressurization. The reactor was then closed and connected to a pump. The pump was then used to add CO2 until the reactor reached the desired polymerization pressure. Following pressurization, the reactor was heated to the desired reaction temperature. All polymerizations were conducted in scCO2 at 241 bar and 80 °C for a total of 24 h. The stirrer bar was set to a stirring rate of 350 rpm. The reactor was then cooled to room temperature (23 °C), and CO2 was vented through the pressure relief valve.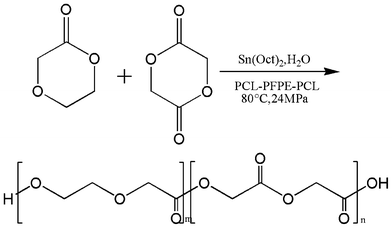 | ||
| Scheme 2 Suspension polymerization of poly(glycolide-co-p-dioxanone) in scCO2. | ||
2.4. Measurements
Microscopic images of particles were obtained from a scanning electron microscope (SEM, FEI INSPECTF) and measured at 10 kV after they were coated with a thin layer of sputtered gold. The particles were ground into liquid nitrogen to observe the fractured surfaces.The particle size and distribution were characterized by a laser diffraction particle size analyzer (Horiba LA-920).
The intrinsic viscosities ([η]) of the resulting polymers were measured in phenol/1,1,2,2–tetrachloroethane (1:1 v/v) solutions using an Ubbelohde viscosimeter thermostated at 25 °C, and all of the solutions were filtered before measurement.
The proton nuclear magnetic resonance (1H-NMR) spectra were recorded on a 400 MHz 1H-NMR spectrometer (Bruker AV-300), using tetramethylsilane as an internal reference and CDCl3 as the solvent for sample 6–7, and HFIP as the solvent for sample 1–5.
DSC analysis was performed on a Phoenix 204 DSC system to study the thermal properties of the synthesized copolymers. The samples were first heated to 230 °C and kept at this temperature for 5 min to eliminate the thermal history. The cooling curves were recorded when the samples were cooled from 230 °C to 30 °C at a nominal rate of 10 °C min−1. After keeping the samples at 30 °C for 5 min, the heating curves were recorded when they were heated from 30 °C to 230 °C at a nominal rate of 10 °C min−1.
X-ray diffractometric analysis was carried out using a diffractometer equipped with a Cu-Kα (λ = 0.154 nm) source, an INEL monochromator, and a goniometric plate.
3. Results and discussion
3.1 Solubilities of the stabilizer and monomers in scCO2
The solubility of this stabilizer were examined by Daniel et al. at 80 °C and 241 bar in scCO2.16 The stabilizer was observed to be soluble under these conditions, demonstrating its CO2-philicity up to loadings of 0.006 g mL−1, which is equivalent to that used during our polymerization reactions in the 100 mL autoclave. However, GA and PDO monomers were found to be insoluble by viewing the cell window under these conditions, as was Sn(Oct)2. Thus, the polymerization of lactide monomers must proceed via a suspension mechanism, which involves the direct conversion of monomer droplets in the continuous phase to solid polymer.3.2 Suspension polymerization of GA and PDO in the presence of the stabilizer
A series of PGDO random copolymers with different compositions were synthesized by suspension polymerization of GA and PDO comonomers (Table 1) with a 15% stabilizer loading at a constant stirring rate of 350 rpm, as illustrated in Scheme 2. The GA to PDO ratio in the resultant copolymers was determined by 1H NMR spectroscopy. Fig. 1 illustrated a typical 1H-NMR spectra of sample 7. The existence of PGA segments can be identified by the chemical shifts at 4.73–4.77 (δHd) and 4.80–4.87 (δHd) ppm, which were attributed to the resonances of –CH3 of PGA. The partly overlapped peaks observed at 3.70–3.82 (δHb), 4.10–4.30 (δHc), 4.33–4.40 (δHa) ppm are attributed to –CH2– of PPDO chains. These results demonstrated that the copolymers are composed of PGA and PPDO segments.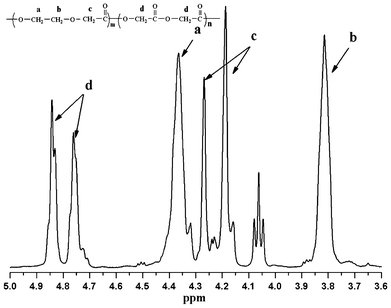
| Samplea | GA (Feed) | GA (NMR) | Yield | [η] | Morphologyf |
|---|---|---|---|---|---|
| (wt%)b | (wt%)c | (%)d | (dl/g)e | ||
|
a Synthesized at 80 °C and 24 MPa for 24 h with 6 g of the monomers, 0.09 mL of Sn(Oct)2, and 15% (w/w) PCL-PFPE-PCL stabilizer.
b Amount of GA in the feedstock.
c Amount of GA units in the polymer chain, as determined by 1H NMR.
d Determined gravimetrically. The yields were estimated for aggregated samples.
e [η] was measured in phenol/1, 1, 2, 2-tetrachloroethane (1:1 v/v) at 25 °C.
f Appearance of the product directly from the autoclave.
|
|||||
| 1 | 100 | 100 | 85 | 0.37 | Fine Powder |
| 2 | 90 | 94.2 | 89 | 0.30 | Fine Powder |
| 3 | 80 | 85.3 | 82 | 0.32 | Fine Powder |
| 4 | 70 | 75.1 | 84 | 0.28 | Fine Powder |
| 5 | 60 | 66.5 | 85 | 0.24 | Fine Powder |
| 6 | 50 | 53.4 | >80 | 0.30 | Aggregated |
| 7 | 40 | 44.9 | >82 | 0.28 | Aggregated |
The intrinsic viscosities and yields (determined gravimetrically) of copolymers obtained can be found in Table 1. The intrinsic viscosities and yield data are acceptable. The intrinsic viscosities of these products range from 0.24 to 0.37. These results reveal that low molecular weights were obtained, and this can be attributed to the presence of hydroxyl impurities in the monomers and a carbonation reaction in scCO2 compared with in conventional solvent.25
3.3 Particle Morphology and size distributions of PGDO with different compositions
The polymerization proceeds via a suspension mechanism, which involves the direct conversion of monomer droplets in the continuous phase to polymer particles. In suspension polymerization, the morphology of the products is known to be sensitive to the rate of stirring and amount of stabilizer added during the reaction.26 So to study how the composition affects the final product morphology, the stirring rate and the content of the stabilizer were kept the same in all experiments. The morphology obtained appears to depend strongly on the copolymer composition. When more than 50% (w/w) GA was fed, the products were obtained as powders (samples 1–5, Table 1; SEM images of samples are shown in Fig. 2). All other compositions resulted in aggregated products which were very similar to that obtained by conventional bulk polymerization at this temperature. | ||
| Fig. 2 SEM images of particles produced by suspension polymerization in scCO2: (a) PGA (100 w/w); (b) PGDO (90/10 w/w); (c) PGDO (80/20 w/w); (d) PGDO (70/30 w/w); (e) PGDO (60/40 w/w). | ||
For the powdery samples, some particles with spherical morphology were contained in the products when an intermediate content of GA between 70% to 90% (w/w) was fed (Fig. 2b, 2d, 2d). In a sharp contrast to the pure PGA sample (Fig. 2a), good control of spherical morphology was observed for the sample fed with 90% (w/w) GA (Fig. 2b). This is a very interesting result that was not reported previously. For suspension polymerization of biodegradable homo- or co-polymers, such as PGA, PLLA, PPDO and PLGA, the morphologies of the produced particles are all irregular.15–19 The irregular morphology limits their applications in many fields.
From Fig. 2, it can also be observed that with a decrease in the GA content from 90% (w/w) to a lower value, the percentage of spherical particles decreased. At the critical point of 60% (w/w), the particles were irregular shapes with coarse surfaces. The magnified photos shown in Fig. 3 further indicated that with this gradual change in particle morphology, serious aggregation between particles occurred. Fig. 4 showed the particle size distributions of samples 1–5. We can see that the mean size of the PGA particles is larger (83.2 μm of sample 1) than the other four samples, and for the samples 2–5, the mean size increased as the content of GA decreased, from 22.4 μm (sample 2) to 61.8 μm (sample 5).
 | ||
| Fig. 3 Magnification photos of particles produced by suspension polymerization in scCO2: (a) PGDO (80/20 w/w); (b) PGDO (70/30 w/w); (c) PGDO (60/40 w/w). | ||
 | ||
Fig. 4 Particle size distributions obtained with ( ) PGA (100 w/w); ( ) PGA (100 w/w); ( ) PGDO (90/10 w/w); ( ) PGDO (90/10 w/w); ( ) PGDO (80/20 w/w); ( ) PGDO (80/20 w/w); ( ) PGDO (70/30 w/w); (▲) PGDO (60/40 w/w). ) PGDO (70/30 w/w); (▲) PGDO (60/40 w/w). | ||
3.4 X-ray diffractometric analysis of PGDO with different compositions
The crystalline structures of the suspension-polymerized samples were investigated by XRD (Fig. 5). In the spectra, the appearance of Bragg peaks at 22.25° and 28.88° correspond to (010) crystal plane and (020) crystal plane for PGA homopolymer. The density of the peak at 22.25° gradually decreased as the PDO content increased. However, there was no obvious change for the peak at 28.88°, indicating the decreased crystallinity of the GA segments with increasing PDO content.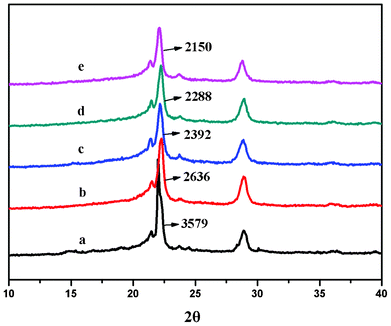 | ||
| Fig. 5 XRD spectra for the PGDO particles acquired in scCO2: (a) PGA (100 w/w); (b) PGDO (90/10 w/w); (c) PGDO (80/20 w/w); (d) PGDO (70/30 w/w); (e) PGDO(60/40 w/w). | ||
3.5 Thermal Analysis of PGDO with different compositions
The series of PGDO copolymers with different compositions were analyzed by DSC (Fig. 6). The DSC second heating curves (performed at 10 °C min−1) of the copolymers are shown in Fig. 6a. The peak melting point of the copolymers is lower than the homopolymer PGA and decreases as the content of PDO increases. However, the peak melting point of sample 2 is even lower than sample 5. This fact is in contrast to the general knowledge that the crystallinity of the copolymers decreased with increasing PDO content. The above anomaly may be ascribed to the much smaller particle size of sample 2 as compared with other crystallized particle samples. Different from the bulk samples, the particle sample with a large surface area contains a very high percentage of less perfect crystals on the surface, leading to a notably decreased melting point. From Fig. 6a, the melting peak is getting weaker at the same time, indicating the significantly decreased crystallinity of the GA segments with increasing PDO content.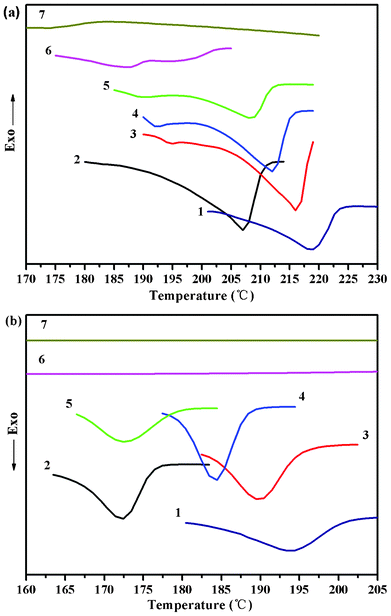 | ||
| Fig. 6 DSC first heating scans (a), subsequent cooling scans (b) of the copolymer samples: (1) PGA (100 w/w); (2) PGDO (90/10 w/w); (3) PGDO (80/20 w/w); (4) PGDO (70/30 w/w); (5) PGDO(60/40 w/w); (6) PGDO(50/50 w/w); (7) PGDO(40/60 w/w); | ||
Fig. 6b shows DSC subsequent cooling scans of the copolymers and equivalent homopolymer from 230 °C. Since the PDO disturbs the regularity of the polymer chains, the crystalline behavior has been changed, which has also been tested by XRD. This is a consequence of the PDO, which greatly changed the microstructure of the copolymers and suppressed the crystallization of the PGA segments during the cooling scans.
3.6 Mechanism for generation of spherical particles
It is well known that scCO2 has the ability to plasticize amorphous polymers. To generate a particulate product via suspension polymerization, the sample must be able to crystallize and be resistant to the plasticizing effect of scCO2.27 For this case, the polymerizing droplets could be effectively solidified by crystallization as the polymerization is complete, leading to the formation of particles. If the polymer is amorphous, it is in a molten state throughout the whole polymerization process due to the plasticization effect of scCO2. At the late stage of polymerization, the polymerizing droplets become very viscous and tend to merge into blocks and precipitate from scCO2. Thus, particulate products could only be acquired from suspension polymerization of crystallized polymers in scCO2.However for most cases, the particles obtained from crystallized biodegradable polymers via suspension polymerization have rough surfaces and irregular shapes. Much attention has been paid to how the reaction conditions affected the size of produced particles, but no control was achieved for the particle morphology. Previously we demonstrated that the PGDO particle morphology was significantly affected by copolymer composition. Considering the simple relation between composition and crystallizability of the copolymers, the crystallization behavior within the polymer/monomers/CO2 mixture droplets is assumed to be the dominant factor to determine the morphology of produced particles.
Previously we have shown that strong or weak crystallizability of the copolymers (sample 1 and sample 5) were not beneficial to acquire particles with well-defined morphology. A tentative explanation for the above two cases is illustrated in Fig. 7 (case 1 and case 3). In contrast to normal suspension polymerization with water as the medium, the polymerizing droplets in scCO2 actually contained three parts, the unreacted monomers, the polymerized product and the dissolved CO2. scCO2 could not only plasticize amorphous polymer, but could also dissolve into the monomers at a certain concentration and changed the phase state of the polymerizing droplets. If the polymer had very strong crystallizability (case 1), it had a high tendency to crystallize during polymerization and precipitated within the droplets. This phase-separation could take place at early stage of polymerization, and profoundly affected the stability of the suspension droplets. The morphology of the resultant particles was thus less controlled. The instability of the polymerizing droplets gave rise to serious aggregation of formed particles, leading to significantly increased particle size as compared with other copolymer particulate products. On the contrary, if the copolymer had weak crystallizability (case 3), the crystallization inside the polymerizing droplets was basically suppressed. Since the plasticizing effect of scCO2 was dominant, the polymerizing droplets could not be fully solidified when the polymerization was completed. During the venting process, the polymerized product was rapidly coagulated due to the release of CO2, but the morphology of the original droplets was also completely disrupted by the turbulence of the gas.
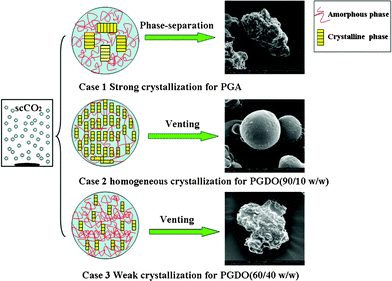 | ||
| Fig. 7 Illustration of particle formation process and morphology by suspension polymerization in scCO2: case 1 polymer has strong crystallizability that leads to serious phase-separation within the polymerizing droplets; case 2 polymer has moderate crystallizability, and the crystallization inside the droplets proceeds homogeneously, leading to the freezing of spherical morphology; case 3 polymer has weak crystallizability, and the droplets could not be effectively solidified due to the plasticizing effect of scCO2. | ||
A balance between the two opposing effects, the crystallization and plasticization of the polymerizing droplets in scCO2, allowed the generation of spherical particles. A typical example is sample 2, for which a delicate balance between the two effects was achieved. The illustration for formation of spherical particles is depicted as case 2 in Fig. 7. For this ideal case, the copolymer had suitable crystallizability and the crystallization did not incur serious phase separation inside the polymerizing droplets. With polymerization, homogeneous crystallization proceeded steadily within the whole region of each single polymerizing droplet. The crystallinity of the polymer was high enough to overcome the plasticization effect of scCO2 and effectively frozen the spherical morphology of original suspension droplets.
In order to confirm the above assumptions, we ground the particles in liquid nitrogen and analyzed the typical fractured surfaces for PGA (100 w/w), PGDO (90/10 w/w) and PGDO (60/40 w/w) three samples by SEM (Fig. 8). For irregular PGA particles, a clear phase-separated structure with large rod-like crystals was found in the interior of the particle (Fig. 8a). The rod-like crystals are very rigid and have smooth surface, indicating the precipitated PGA crystals within the droplet. In contrast, for spherical PGDO (90/10 w/w) particles, the cross region of the particle has a relatively homogeneous texture with very uniform distribution of tiny crystal grains. That means the crystallization inside the droplet occurred very uniformly and evenly at a controlled manner. From Fig. 8c, the irregular particle is composed of multiple un-solidified droplets that were deformed, stretched and coagulated during the venting process. The SEM cross pictures for the particles are very consistent with above mentioned three cases used to explain the particle formation process and morphology.
 | ||
| Fig. 8 SEM fractured surfaces of the particles: (a) PGA (100 w/w); (b) PGDO (90/10 w/w); (c) PGDO(60/40 w/w). | ||
4. Conclusion
PGDO copolymers with different compositions were synthesized by suspension ring-opening polymerization of GA and PDO monomers using Sn(Oct)2 as a catalyst and a fluorocarbon polymer surfactant as a stabilizer. The molecular weight and yield data are acceptable. It was found that to produce PGDO particles, the copolymer must be semicrystalline, which means a sample of PGDO must contain more than 50% (w/w) GA. At the same time, the mean diameter of particles greatly increases as the content of PDO increases. This is the first report that spherical PGDO microparticles were obtained when 70% to 90% (w/w) GA was fed. The result is very consistent with the mechanism of suspension polymerization, where the PGDO is soluble in its GA monomer in the presence of scCO2. This work will have more applications in pharmaceutical areas.Acknowledgements
This work was supported by the National Natural Sciences Fund of China (No. 30970725 and No. 51273034), and the Fundamental Research Funds for the Central Universities.References
- L. T. Lim, R. Auras and M. Rubino, Prog. Polym. Sci., 2008, 33, 820 CrossRef CAS.
- R. Chandra and R. Rustgi, Prog. Polym. Sci., 1998, 23, 1273 CrossRef CAS.
- D. Kato, M. Takeuchi, T. Sakurai, S. Furukawa, H. Mizokami, M. Sakata, C. Hirayama and M. Kunitake, Biomaterials, 2003, 24, 4253 CrossRef CAS.
- H. W. Kim, H. J. Gu and H. H. Lee, Tissue Eng., 2007, 13, 965 CrossRef CAS.
- K. Mishima, Adv. Drug Delivery Rev., 2008, 60, 411 CrossRef CAS.
- A. Martín and M. J. Cocero, Adv. Drug Delivery Rev., 2008, 60, 339 CrossRef.
- K. A. Shaffer and J. M. DeSimone, Trends Polym Sci., 1995, 3, 146 CAS.
- D. A. Canelas and J. M. DeSimone, Adv. Polym. Sci., 1997, 133, 103 CrossRef CAS.
- J. Jung and M. Perrut, J. Supercrit. Fluids, 2001, 20, 179 CrossRef CAS.
- N. Yildiz, S. Tuna, O. Döer and A. Calimli, J. Supercrit. Fluids, 2007, 41, 440 CrossRef CAS.
- J. Y. Hao, M. J. Whitaker, G. Serhatkulu, K. M. Shakesheff and S. M. Howdle, J. Mater. Chem., 2005, 15, 1148 RSC.
- M. Strumendo, A. Bwetucco and N. Elvassore, J. Supercrit. Fluids, 2007, 41, 115 CrossRef CAS.
- J. W. Pack, S. H. Kim, S. Y. Pack, Y. W. Lee and Y. H. Kim, Macromol. Biosci., 2004, 4, 340 CrossRef CAS.
- E. Reverchon, G. D. Porta, A. Di Trolio and S. Pace, Ind. Eng. Chem. Res., 1998, 37, 952 CrossRef CAS.
- D. Bratton, M. Brown and S. M. Howdle, Chem. Commun., 2004, 808 RSC.
- D. Bratton, M. Brown and S. M. Howdle, Macromolecules, 2003, 36, 5908 CrossRef CAS.
- H. S. Ganapathy, H. S. Hwang, Y. T. Jeong, W. K. Lee and K. T. Lim, Eur. Polym. J., 2007, 43, 119 CrossRef CAS.
- T. Q. Wang, X. L. Zhao and J. Y. Hao, Chin. Chem. Lett., 2011, 22, 1509 CrossRef CAS.
- D. Bratton, M. Brown and S. M. Howdle, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6573 CrossRef CAS.
- B. Grignard, F. Stassin, C. Calberg, R. Jérôme and C. Jérôme, Biomacromolecules, 2008, 9, 3141 CrossRef CAS.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Chem. Rev., 1999, 99, 543 CrossRef CAS.
- US Pat., 5391707, 1995 Search PubMed.
- F. Pilati, M. Toselli, M. Messori, A. Priola, R. Bongiovanni, G. Malucellia and C. Tonelli, Macromolecules, 1999, 32, 6969 CrossRef CAS.
- A. Duda, S. Penczek, A. Kowaski and J. Libiszowski, Macromol. Symp., 2000, 153, 41 CrossRef CAS.
- F. Stassin, O. Halleux and R. Jérôme, Macromolecules, 2001, 34, 775 CrossRef CAS.
- H.G. Yuan, G. Kalfas and W.H. Ray, J. Macromol. Sci., Part C, 1991, C31 (2–3), 215 CrossRef.
- D. L. Tomasko, H. Li, D. Liu, X. Han, M. J. Wingert, L. J. Lee and K. W. Koelling, Ind. Eng. Chem. Res., 2003, 42, 6431 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |